
Cellular Therapy Updates in B-Cell Lymphoma: The State of the CAR-T


Abstract
:Simple Summary
Abstract
1. Introduction
2. US FDA-Approved CAR-T Therapies for B-Cell Lymphomas
2.1. Axicabtagene Ciloleucel
2.1.1. CAR Construct
2.1.2. Efficacy
2.1.3. Toxicity
2.2. Tisagenlecleucel
2.2.1. CAR Construct
2.2.2. Efficacy
2.2.3. Toxicity
2.3. Brexucabtagene Autoleucel
2.3.1. CAR Construct
2.3.2. Efficacy
2.3.3. Toxicity
2.4. Lisocabtagene Maraleucel
2.4.1. CAR Construct
2.4.2. Efficacy
2.4.3. Toxicity
3. Promising Cellular Therapy Research for B-Cell Lymphomas
3.1. Targeting New Antigens
3.2. Multi-Specific CARs
3.3. Allogeneic “Off the Shelf” CAR-Ts
3.4. Optimizing CAR-T Signaling, Expansion and Persistence
3.5. Reducing CAR-T Toxicity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Neste, E.V.D.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Shankland, K.R.; Armitage, J.O.; Hancock, B.W. Non-Hodgkin lymphoma. Lancet 2012, 380, 848–857. [Google Scholar] [CrossRef]
- Pileri, S.A.; Milani, M.; Orcioni, G.F.; Sabattini, E. From the R.E.A.L. Classification to the upcoming WHO scheme: A step toward universal categorization of lymphoma entities? Ann. Oncol. 1998, 9, 607–612. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J. Lymphoma classification – from controversy to consensus: The R.E.A.L. and WHO Classification of lymphoid neoplasms. Ann. Oncol. 2000, 11, S3–S10. [Google Scholar] [CrossRef]
- Bartlett, N.; Wilson, W.H.; Jung, S.-H.; Hsi, E.D.; Maurer, M.J.; Pederson, L.D.; Polley, M.-Y.C.; Pitcher, B.N.; Cheson, B.D.; Kahl, B.S.; et al. Dose-Adjusted EPOCH-R Compared With R-CHOP as Frontline Therapy for Diffuse Large B-Cell Lymphoma: Clinical Outcomes of the Phase III Intergroup Trial Alliance/CALGB 50303. J. Clin. Oncol. 2019, 37, 1790–1799. [Google Scholar] [CrossRef]
- Sehn, L.H.; Berry, B.; Chhanabhai, M.; Fitzgerald, C.; Gill, K.; Hoskins, P.; Klasa, R.; Savage, K.J.; Shenkier, T.; Sutherland, J.; et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood 2007, 109, 1857–1861. [Google Scholar] [CrossRef] [Green Version]
- Gisselbrecht, C.; Glass, B.; Mounier, N.; Gill, D.S.; Linch, D.C.; Trneny, M.; Bosly, A.; Ketterer, N.; Shpilberg, O.; Hagberg, H.; et al. Salvage Regimens With Autologous Transplantation for Relapsed Large B-Cell Lymphoma in the Rituximab Era. J. Clin. Oncol. 2010, 28, 4184–4190. [Google Scholar] [CrossRef] [Green Version]
- Neste, E.V.D.; Schmitz, N.; Mounier, N.; Gill, D.; Linch, D.; Trneny, M.; Milpied, N.; Radford, J.; Ketterer, N.; Shpilberg, O.; et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the International CORAL study. Bone Marrow Transplant. 2016, 51, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Nagle, S.J.; Woo, K.; Schuster, S.J.; Nasta, S.D.; Stadtmauer, E.; Mick, R.; Svoboda, J. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am. J. Hematol. 2013, 88, 890–894. [Google Scholar] [CrossRef]
- Hamadani, M.; Hari, P.; Zhang, Y.; Carreras, J.; Akpek, G.; Aljurf, M.D.; Ayala, E.; Bachanova, V.; Chen, A.I.; Chen, Y.-B.; et al. Early Failure of Frontline Rituximab-Containing Chemo-immunotherapy in Diffuse Large B Cell Lymphoma Does Not Predict Futility of Autologous Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2014, 20, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Radford, J.; Davies, A.; Cartron, G.; Morschhauser, F.; Salles, G.; Marcus, R.; Wenger, M.; Lei, G.; Wassner-Fritsch, E.; Vitolo, U. Obinutuzumab (GA101) plus CHOP or FC in relapsed/refractory follicular lymphoma: Results of the GAUDI study (BO21000). Blood 2013, 122, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, A.; Hoster, E.; Zwingers, T.; Brittinger, G.; Engelhard, M.; Meusers, P.; Reiser, M.; Forstpointner, R.; Metzner, B.; Peter, N.; et al. Improvement of Overall Survival in Advanced Stage Mantle Cell Lymphoma. J. Clin. Oncol. 2009, 27, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoster, E.; Dreyling, M.; Klapper, W.; Gisselbrecht, C.; Van Hoof, A.; Kluin-Nelemans, J.C.; Pfreundschuh, M.; Reiser, M.; Metzner, B.; Einsele, H.; et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Tiemann, M.; Schrader, C.; Klapper, W.; Dreyling, M.H.; Campo, E.; Norton, A.; Berger, F.; Kluin, P.; Ott, G.; Pileri, S.; et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): A clinicopathological study from the European MCL Network. Br. J. Haematol. 2005, 131, 29–38. [Google Scholar] [CrossRef]
- Majlis, A.; Pugh, W.C.; Rodriguez, M.A.; Benedict, W.F.; Cabanillas, F. Mantle cell lymphoma: Correlation of clinical outcome and biologic features with three histologic variants. J. Clin. Oncol. 1997, 15, 1664–1671. [Google Scholar] [CrossRef]
- Ferrero, S.; Rossi, D.; Rinaldi, A.; Bruscaggin, A.; Spina, V.; Eskelund, C.W.; Evangelista, A.; Moia, R.; Kwee, I.; Dahl, C.; et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: A FIL study. Haematologica 2019, 105, 1604–1612. [Google Scholar] [CrossRef] [Green Version]
- Rummel, M.J.; Niederle, N.; Maschmeyer, G.; Banat, G.A.; von Grünhagen, U.; Losem, C.; Kofahl-Krause, D.; Heil, G.; Welslau, M.; Balser, C.; et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013, 381, 1203–1210. [Google Scholar] [CrossRef]
- Flinn, I.W.; Van Der Jagt, R.; Kahl, B.; Wood, P.; Hawkins, T.; Macdonald, D.; Simpson, D.; Kolibaba, K.; Issa, S.; Chang, J.; et al. First-Line Treatment of Patients with Indolent Non-Hodgkin Lymphoma or Mantle-Cell Lymphoma With Bendamustine Plus Rituximab Versus R-CHOP or R-CVP: Results of the BRIGHT 5-Year Follow-Up Study. J. Clin. Oncol. 2019, 37, 984–991. [Google Scholar] [CrossRef]
- Romaguera, J.E.; Fayad, L.; Rodriguez, M.A.; Broglio, K.R.; Hagemeister, F.B.; Pro, B.; McLaughlin, P.; Younes, A.; Samaniego, F.; Goy, A.; et al. High Rate of Durable Remissions After Treatment of Newly Diagnosed Aggressive Mantle-Cell Lymphoma with Rituximab Plus Hyper-CVAD Alternating with Rituximab Plus High-Dose Methotrexate and Cytarabine. J. Clin. Oncol. 2005, 23, 7013–7023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Nobel Prize in Physiology or Medicine 1908. Available online: https://www.nobelprize.org/prizes/medicine/1908/summary/ (accessed on 8 August 2021).
- Allison, J.; McIntyre, B.W.; Bloch, D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 1982, 129, 2293–2300. [Google Scholar] [PubMed]
- Kuhns, M.S.; Badgandi, H.B. Piecing together the family portrait of TCR-CD3 complexes. Immunol. Rev. 2012, 250, 120–143. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.-H.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Aebersold, P.; Cornetta, K.; Kasid, A.; Morgan, R.A.; Moen, R.; Karson, E.M.; Lotze, M.T.; Yang, J.C.; Topalian, S.L.; et al. Gene Transfer into Humans — Immunotherapy of Patients with Advanced Melanoma, Using Tumor-Infiltrating Lymphocytes Modified by Retroviral Gene Transduction. N. Engl. J. Med. 1990, 323, 570–578. [Google Scholar] [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Jain, M.; Feng, L.; Spiegel, J.Y.; Ghobadi, A.; Lin, Y.; Dahiya, S.; Lunning, M.; Lekakis, L.; Reagan, P.; et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results from the US Lymphoma CAR T Consortium. J. Clin. Oncol. 2020, 38, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.C.; Chavez, J.C.; Sehgal, A.R.; William, M.B.M.; Munoz, J.; Salles, M.G.; Munshi, P.N.; Casulo, C.; Maloney, D.; de Vos, S.; et al. Primary Analysis of Zuma-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-Hodgkin Lymphoma (iNHL). Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Jaeger, U. Myc Expression and Tumor-Infiltrating T Cells Are Associated with Response in Patients (Pts) with Relapsed/Refractory Diffuse Large B-Cell Lymphoma (r/r DLBCL) Treated with Tisagenlecleucel in the Juliet Trial. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2021; American Society of Hematology: Washington, DC, USA, 2020. [Google Scholar]
- Chong, E.A.; Ruella, M.; Schuster, S.J. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N. Engl. J. Med. 2021, 384, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, M.C.; Hu, Z.-H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Upadhaya, S.; Yu, J.X.; Shah, M.; Correa, D.; Partridge, T.; Campbell, J. The clinical pipeline for cancer cell therapies. Nat. Rev. Drug Discov. 2021, 20, 503–504. [Google Scholar] [CrossRef]
- Yang, S.; Li, J.-Y.; Xu, W. Role of BAFF/BAFF-R axis in B-cell non-Hodgkin lymphoma. Crit. Rev. Oncol. 2014, 91, 113–122. [Google Scholar] [CrossRef]
- Qin, H.; Dong, Z.; Wang, X.; Cheng, W.A.; Wen, F.; Xue, W.; Sun, H.; Walter, M.; Wei, G.; Smith, D.L.; et al. CAR T cells targeting BAFF-R can overcome CD19 antigen loss in B cell malignancies. Sci. Transl. Med. 2019, 11, eaaw9414. [Google Scholar] [CrossRef]
- Ormhøj, M.; Scarfò, I.; Cabral, M.L.; Bailey, S.; Lorrey, S.J.; Bouffard, A.A.; Castano, A.P.; Larson, R.C.; Riley, L.S.; Schmidts, A.; et al. Chimeric Antigen Receptor T Cells Targeting CD79b Show Efficacy in Lymphoma with or without Cotargeting CD19. Clin. Cancer Res. 2019, 25, 7046–7057. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Köksal, H.; Dillard, P.; Josefsson, S.E.; Maggadottir, S.M.; Pollmann, S.; Fåne, A.; Blaker, Y.N.; Beiske, K.; Huse, K.; Kolstad, A.; et al. Preclinical development of CD37CAR T-cell therapy for treatment of B-cell lymphoma. Blood Adv. 2019, 3, 1230–1243. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Ghafouri, S.N.; Walthers, C.; Roshandell, M.; Ji, B.; Trent, J.; Chen, J.M.; Naparstek, J.; Harris, C.; Schweppe, T.; Nawaly, K.K.; et al. Abstract CT007: CD19/CD20 Bispecific Chimeric Antigen Receptor (CAR) in Naive/Memory T-Cells for the Treatment of Relapsed or Refractory B-Cell Lymphomas. In Clinical Trials; American Association for Cancer Research: Philadelphia, PA, USA, 2021; p. CT007. [Google Scholar]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–10. [Google Scholar] [CrossRef]
- Hossain, N.; Sahaf, B.; Abramian, M.; Spiegel, J.Y.; Kong, K.; Kim, S.; Mavroukakis, S.; Oak, J.; Natkunam, Y.; Meyer, E.H.; et al. Phase I Experience with a Bi-Specific CAR Targeting CD19 and CD22 in Adults with B-Cell Malignancies. Blood 2018, 132, 490. [Google Scholar] [CrossRef]
- Cronk, R.J.; Zurko, J.; Shah, N.N. Bispecific Chimeric Antigen Receptor T Cell Therapy for B Cell Malignancies and Multiple Myeloma. Cancers 2020, 12, 2523. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiadis, C.; Preece, R.; Nickolay, L.; Etuk, A.; Petrova, A.; Ladon, D.; Danyi, A.; Humphryes-Kirilov, N.; Ajetunmobi, A.; Kim, D.; et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol. Ther. 2018, 26, 1215–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPersio, J.F.; Staser, K.; Cooper, M. Immunotherapy for T-Cell ALL and T-Cell NHL. Clin. Lymphoma Myeloma Leuk. 2020, 20, S56–S58. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef]
- Perez, C.; Gruber, I.; Arber, C. Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring “Universal” Donor T Cells. Front. Immunol. 2020, 11, 583716. [Google Scholar] [CrossRef]
- Ayyappan, S.; Maddocks, K. Novel and emerging therapies for B cell lymphoma. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Wen, Q.; Zhong, J.F.; Zhang, C.; Zhang, X. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Qin, L.; Zhao, R.; Chen, D.; Wei, X.; Wu, Q.; Long, Y.; Jiang, Z.; Li, Y.; Wu, H.; Zhang, X.; et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark. Res. 2020, 8, 1–12. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Chong, E.A.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Winchell, N.; Napier, E.; Mato, A.R.; Melenhorst, J.J.; Ruella, M.; Lacey, S.F.; et al. Sequential Anti-CD19 Directed Chimeric Antigen Receptor Modified T-Cell Therapy (CART19) and PD-1 Blockade with Pembrolizumab in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphomas. Blood 2018, 132, 4198. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Locke, F.L.; Miklos, D.B.; Herrera, A.F.; Westin, J.R.; Lee, J.; Rossi, J.M.; Zheng, L.; Avanzi, M.P.; Roberts, Z.J.; et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Blood 2018, 132, 4192. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. Switching CAR-T cells on and off. Nat. Rev. Drug Discov. 2021, 20, 100. [Google Scholar] [CrossRef] [PubMed]
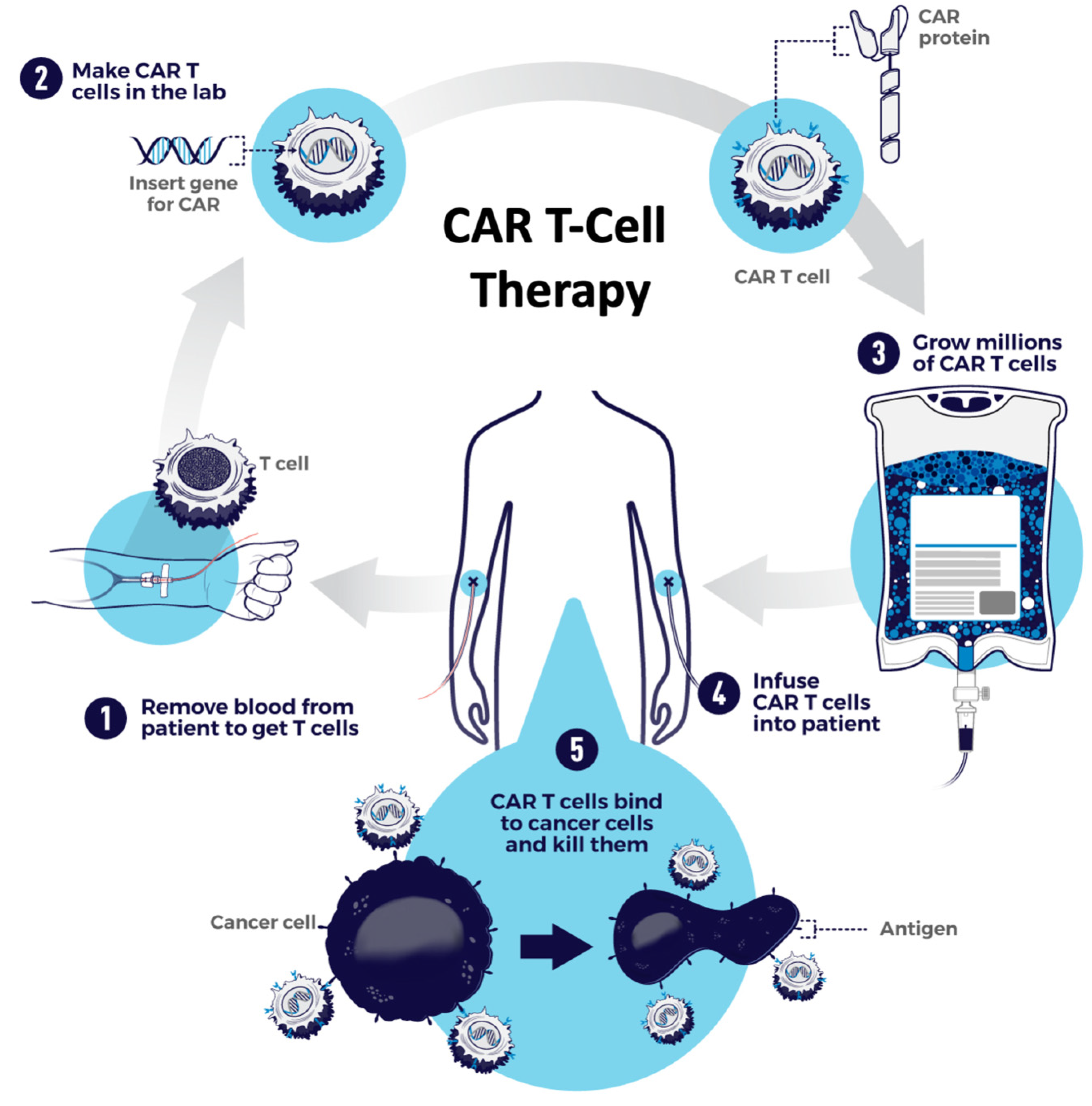




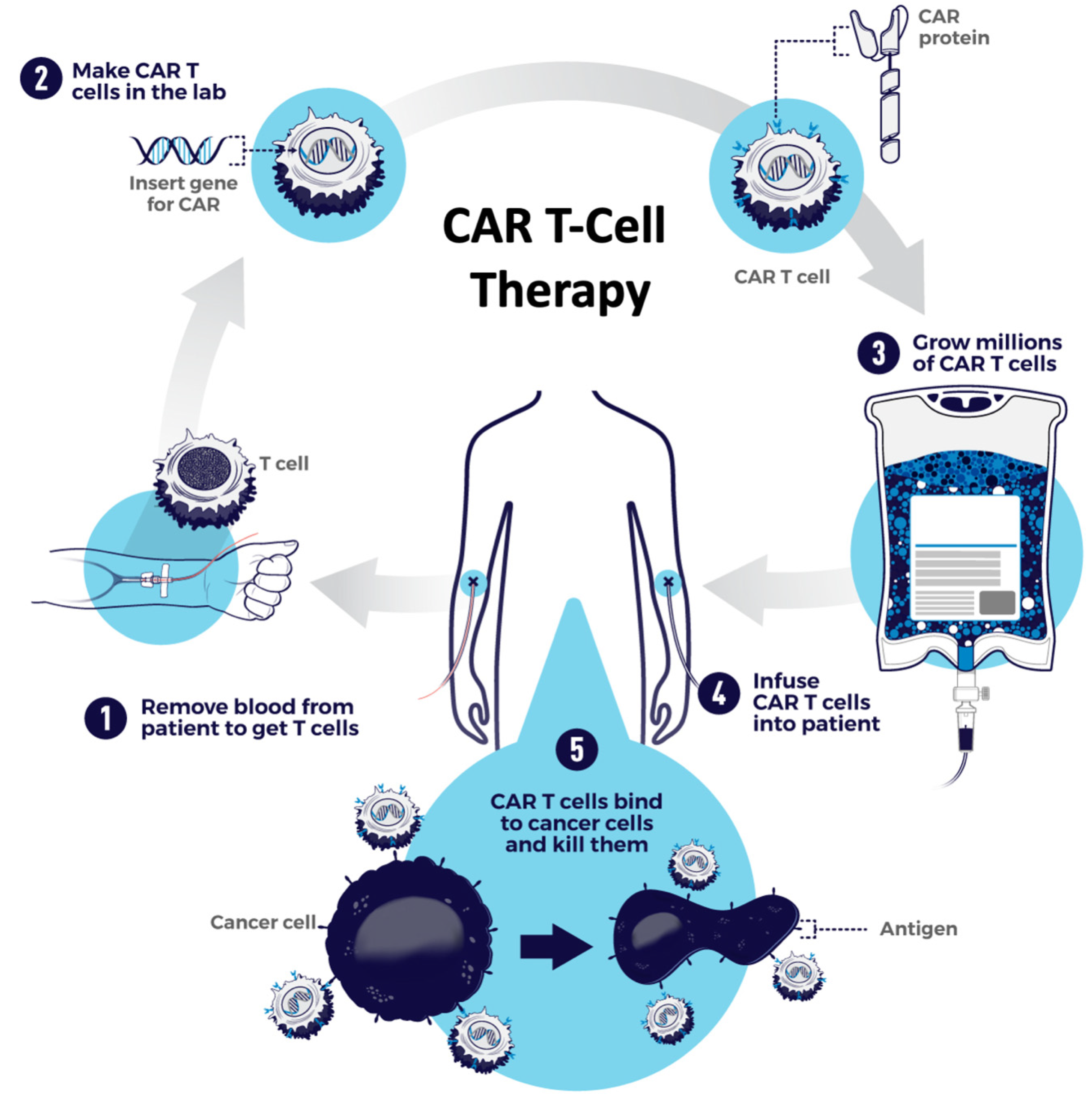{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Axicabtagene Ciloleucel (axi-cel, Yescarta) | Tisagenlecleucel (tisa-cel, Kymriah) | Brexucabtagene Autoleucel (brexu-cel, Tecartus) | Lisocabtagene Maraleucel (liso-cel, Breyanzi) | |
|---|---|---|---|---|
| US FDA Approval | 2017: LBCL 2021: FL | 2017: ALL (age < 26 years) 2018: LBCL | 2020: MCL | 2021: LBCL |
| CAR Construct | •2nd gen, CD28 •Retroviral vector | •2nd gen, 41BB •Lentiviral vector | •2nd gen, CD28 •Retroviral vector | •2nd gen, 41BB •Lentiviral vector |
| Pivotal Trial | ZUMA-1 [4] Phase 1/2 | JULIET [5] Phase 2 | ZUMA-2 [6] Phase 2 | TRANSCEND [7] Phase 2 |
| Study Population | 111 enrolled; 101 dosed •76% DLBCL; 16% tFL; 8% PMBCL •79% refractory •21% post-ASCT | 165 enrolled; 111 dosed •80% DLBCL; 18% tFL •54% refractory •49% post-ASCT | 74 enrolled, 68 dosed •100% MCL •40% refractory •43% post-ASCT | 344 enrolled, 269 dosed •51% DLBCL, 13% HGBCL, 6% PMBCL, 1% FL grade 3b •67% refractory •35% post-ASCT |
| Bridging Chemo | No | Yes, 59% | Yes, 37% | Yes, 59% |
| Lymphodepleting Chemo | Flu 30 mg/m2 and Cy 500 mg/m2 on Days −5, − 4, and − 3 | •Flu 25 mg/m2 and Cy 250 mg/m2 on Days −5, − 4, and − 3 •Bendamustine (90 mg/m2) daily for 2 days | Flu 30 mg/m2 and Cy 500 mg/m2 on Days −5, − 4, and − 3 | Flu 30 mg/m2 and Cy 300 mg/m2 × 3 days, 2–7 days before CAR infusion |
| CAR-T Dose | •2.0 × 106 CAR-T cells/kg •If >100 kg, max. 2.0 × 108 CAR-T cells | •Median, 3 × 108 CAR-T cells •Range, 0.1–6.0 × 108 cells | •2.0 × 106 CAR-T cells/kg •If >100 kg, max. 2.0 × 108 CAR-T cells) | •DL1: 50 × 106 CAR-T cells (n = 45) •DL1: 100 × 106 CAR-T cells (n = 183) •DL3: 150 × 106 CAR-T cells (n = 41) (CD4:CD8 in 1:1 ratio) |
| Efficacy | OR: 82% CR: 54% Med. DOR: 11.1 mo Med. PFS: 5.9 mo OS at 18 mo: 52% | OR: 52% CR: 40% Med. DOR: NR at 17 mo Med. OS: 11.1 months | OR: 85% CR: 59% Med. DOR: NR at 12 mo PFS at 12 mo: 61% OS at 12 mo: 83% | OR: 61% CR: 44% Med. DOR: NR at 12 mo Med. PFS: 6.8 mo Med. OS: 21.1 mo |
| Safety | CRS: •All Grades: 93% •≥ Grade 3: 13% Neurotoxicity: •All Grades: 64% •≥ Grade 3: 28% Grade 5 AEs: 6% | CRS: •All Grades: 58% •≥ Grade 3: 22% Neurotoxicity: •All Grades: 21% •≥ Grade 3: 12% Grade 5 AEs: 3% | CRS: •All Grades: 91% •≥ Grade 3: 15% Neurotoxicity: •All Grades: 63% •≥ Grade 3: 31% Grade 5 AEs: 3% | CRS: •All Grade: 42% •≥ Grade 3: 2% Neurotoxicity: •All Grades: 30% •≥ Grade 3: 10% Grade 5 AEs: 0% |
| Future Directions in Translational and Clinical CAR-T Development | |
|---|---|
| Targeting New Antigens | CD20, CD22, BAFF-R, CD29a, CD37, Ig-kappa and others |
| Multi-specific CARs | Co-administration of ≥2 antigen-specific CAR-Ts; Bi-cistronic CAR-Ts expressing ≥2 different antigen-specific CARs; Tandem CAR-Ts expressing 1 CAR with dual antigen specificity (furthest in clinical development: CD19/CD20 and CD19/CD22) |
| Allogeneic “off-the-shelf” Universal CAR-Ts | TCR and/or HLA modifications using various gene editing techniques (e.g., zinc finger nucleases, TALENs and CRISPR-Cas9) |
| Optimizing CAR-T Function | CAR modification (3rd and 4th Gen CAR-Ts), cytokine stimulation/priming (e.g., IL-7), checkpoint inhibition (e.g., PD-1/PD-L1) |
| Reducing CAR-T Toxicity | Prophylactic tocilizumab and/or glucocorticoids, CAR modification, CAR-T “on/off” switches, CAR-T “suicide” switches, targeted inhibitors of relevant inflammatory signaling pathways (e.g., JAK/STAT) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crees, Z.D.; Ghobadi, A. Cellular Therapy Updates in B-Cell Lymphoma: The State of the CAR-T. Cancers 2021, 13, 5181. https://doi.org/10.3390/cancers13205181
Crees ZD, Ghobadi A. Cellular Therapy Updates in B-Cell Lymphoma: The State of the CAR-T. Cancers. 2021; 13(20):5181. https://doi.org/10.3390/cancers13205181
Chicago/Turabian StyleCrees, Zachary D., and Armin Ghobadi. 2021. "Cellular Therapy Updates in B-Cell Lymphoma: The State of the CAR-T" Cancers 13, no. 20: 5181. https://doi.org/10.3390/cancers13205181
APA StyleCrees, Z. D., & Ghobadi, A. (2021). Cellular Therapy Updates in B-Cell Lymphoma: The State of the CAR-T. Cancers, 13(20), 5181. https://doi.org/10.3390/cancers13205181