
The Risk of Malignancies in Celiac Disease—A Literature Review

 ,
,  , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
3. Celiac Disease and the Overall Risk of Malignancies
4. Celiac Disease and Lymphomas
4.1. Enteropathy-Associated T-Cell Lymphoma
4.1.1. Clinical Presentation
4.1.2. Staging and Prognostic Systems
4.1.3. Imaging and Endoscopy
4.1.4. Pathology
4.1.5. Genetics
4.1.6. Treatment

{kind=link}
| Study | Year | Design | No. EATL | No. ASCT | Debulking Chemotherapy | Conditioning Treatment | Response and Survival |
|---|---|---|---|---|---|---|---|
| Gale et al. [89] | 2000 | Retrospective | 31 | 2 | PEACE-BOM | BEAM | 1 CR; disease-free 64 months after diagnosis |
| Blystad et al. [149] | 2001 | Retrospective | 2 (total 40 NHL) | 2 | CHOP, MACOP-B | BEAM (or BEAM-like) + TBI | NR |
| Okuda et al. [150] | 2002 | Retrospective | 1 | 1 | CHOP, ESHAP | MCVC | Relapse after 8 months, death 17 months after ASCT |
| Chonabayashi et al. [151] | 2007 | Retrospective | 1 | 1 | EPOCH-ICE | MPH-FARA + TBI | Alive at 11 months |
| Jantunen et al. [152] | 2003 | Retrospective | 5 | 5 | CHOP | BEAM (or BEAM-like) | OS: 2 months. 2 pts died from TRC; 2 progressed early (0 and 1 month); 1 relapsed and died at 14 months |
| Rongey et al. [153] | 2006 | Retrospective | 1 | 1 | CHOP | BEAM | Alive and in remission at 18 months |
| Bishton et al. [101] | 2007 | Retrospective | 6 | 6 | IVE + HDMTX | BEAM | 5 CR 4 patients alive and in CR after 1.8–4.3 years |
| Al-Toma et al. [154] | 2007 | Retrospective | 4 | 4 | CHOP | BEAM or MPH-FARA | 1 in ongoing CR after 32 months 3 patients died after 2–9 months |
| Nava et al. [155] | 2007 | Retrospective | 1 | 1 | CHOEP | BEAM | No evidence of residual disease 70 days after ASCT |
| Reimer et al. [156] | 2009 | Prospective | 5 (total 83 NHL) | 5 | CHOP + DACMEMP (or PAEM) | TBI + HDCTX | NR |
| Sieniawski et al. [137] | 2010 | Prospective | 54 | 14 | CHOP—IVE/MTX | TBI + HDMPH or BEAM | Remission rate: 69% Death rate: 39% 5-year OS: 60% |
| Prochazka et al. [157] | 2011 | Retrospective | 2 (total 29 NHL) | 2 | PACEB-IVAM-HAM | BEAM | 1 CR; survival not reported |
| Nijeboer et al. [158] | 2015 | Retrospective | 61 | 8 † | Different regimens | MPH-FARA or CTX-FARA | Complete response: 39% Median OS: 7.4 months 5-year OS: 11% |
4.2. Other Lymphomas
5. Celiac Disease and Small Bowel Carcinoma
5.1. Epidemiology
| Authors | Year | Country | Design | Size of the CeD Cohort | No. of SBC | Results |
|---|---|---|---|---|---|---|
| Swinson et al. [17] | 1983 | United Kingdom | Retrospective | 235 | 19 | Included only CeD patients with a diagnosed malignancy. Observed cases: 19; expected cases: 0.23. Relative risk = 82.6 Individual risk is extremely low (50 per 100,000/year) |
| Cottone et al. [50] | 1999 | Italy | Retrospective | 216 | 1 duodenal adenocarcinoma | NR |
| Askling et al. [23] | 2002 | Sweden | Retrospective | Inpatient diagnosed with: CeD 11,019; DH 1354; both diagnoses 226 | 8 cases in CeD cohort (6 adenocarcinomas, 1 mixed carcinoid-adenocarcinoma and 1 unclassified); no cases in DH cohort; 1 case in CeD + DH cohort | Increased risk in CeD cohort: SIR = 10 (95% CI 4.4–20) No significant increased risk in CeD + DH cohort: SIR = 16 (95% CI 0.4–88) |
| Green et al. [29] | 2003 | USA | Prospective | 381 | 3 | Cancer diagnosed before or simultaneously CeD Expected cases: 0.1; SMR = 34 (95% CI 24–42) |
| Card et al. [24] | 2004 | United Kingdom | Prospective | 865 | 1 | Increased risk in the peridiagnostic period (≤2 years after diagnosis of CeD): Crude risk 62 cases/100,000 SIR = 59.97 (95% CI 1.52–334.12) No cases registered in the post diagnostic period (>2 years after diagnosis of CeD) |
| West et al. [25] | 2004 | United Kingdom | Retrospective | 4732 (23,620 controls) | 29 † | Increased risk of gastrointestinal cancers: Overall: aHR = 1.95 (1.27–3.00) First year after diagnosis: aHR = 3.31 (1.40–7.83) Beyond the first year after diagnosis: aHR = 1.65 (0.99–2.76) |
| Silano et al. [26] | 2007 | Italy | Prospective | 1968 | 5 | Cancer diagnosis preceded diagnosis of CeD. Expected cases: 0.19; SIR = 25 (95% CI 8.5–51.4) |
| Anderson et al. [30] | 2007 | United Kingdom | Retrospective | 2079 (490 EMA+; 1133 AGA+; 456 AGA+ and EMA-) | 3 | Despite the increased SIR, no significant association: EMA+: SIR = 23.33 (95% CI 0.00–69.07) AGA+: SIR = 7.28 (95% CI 0.00–21.54) AGA+ and EMA-: SIR = 15.51 (95% CI 0.00–45.90) |
| Lohi et al. [32] | 2009 | Finland | Retrospective | 6849 (202 tTG positive; 73 EMA positive) | 121 † | 115 cases in tTG-negative, 6 cases in tTG-positive, 0 cases in EMA-positive. Relative risk for tTG positive patients = 1.38 (95% CI 0.60–3.14) |
| Grainge et al. [27] | 2012 | United Kingdom | Retrospective | 435 | 1 | Expected cases: 0.09; SIR = 11.1 (95% CI 0.28–61.6) |
| Elfstrom et al. [168] | 2012 | Sweden | Prospective | 28,882 Marsh score 3 12,860 Marsh score 1–2 3705 positive serology | 25 in Marsh 3 24 in Marsh 1–2 4 in positive serology | After 1 year of follow-up after CeD diagnosis: Marsh 3: HR = 2.22 (95% CI 1.19–4.14) Marsh 1–2: HR = 2.49 (1.07–5.79) Positive serology: HR = 4.67 (95% CI 0.53–41.4) |
| Ilus et al. [56] | 2014 | Finland | Retrospective | 32,439 | 27 | Increased risk of SBC All cases: SIR = 4.29 (95% CI 2.83–6.24) Males: SIR = 3.47 (95% CI 1.66–6.37) Females: SIR = 5.00 (95% CI 2.91–7.99) |
| van Gils et al. [49] | 2018 | Netherlands | Retrospective case–control | 261/301,337 cases 282/576,971 controls | 136 with CeD 5335 without CeD | Increased risk of SBC: RR = 11.9 (95% CI 8.2–17.2) |
| Caio et al. [177] | 2019 | Italy | Retrospective | 770 | 5 | NR |
| Emilsson et al. [169] | 2020 | Sweden | Retrospective | 48,119 CeD patients and 239,249 controls | 74 | Beginning 1 year after diagnosis of CeD, 29 CeD patients (0.06%) and 45 controls (0.02%) developed SBC. HR = 3.05 (95% CI 1.86–4.99) 1 extra case of SBC in every 2944 CeD patients followed for 10 years |
5.2. Histopathology, Molecular Biology and Pathogenesis
5.3. Clinical Presentation and Diagnosis
5.4. Prognosis and Treatment
6. Celiac Disease and Other Malignancies
| Study | Year | Country | Study Design | CeD Cases—n | Cancer Cases—n | Main Findings |
|---|---|---|---|---|---|---|
| Colon and rectum cancer | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 26 | Compared to the general population: Increased risk of colon cancer: SIR = 1.9 (95% CI 1.2–2.8) Similar risk of rectum cancer: SIR = 0.8 (95% CI 0.3–1.6) |
| Green et al. [29] | 2003 | USA | Hospital-based prospective cohort study | 381 | 3 | No increased risk of colon cancer: SIR = 0.8 (95% CI 0.1–7.2) |
| Viljamaa et al. [11] | 2006 | Finland | Population-based prospective cohort study | 781 | 4 | No increased risk of colon and rectum cancer: SIR = 1.1 (95% CI 0.3–2.8) |
| Silano et al. [26] | 2007 | Italy | Hospital-based prospective cohort study | 1968 | 7 | No increased risk of colon cancer: SIR = 1.1 (95% CI 0.68–1.56) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 11 colon cancers 4 rectum cancers | No increased risk (excluding cases occurred within the first year after CeD diagnosis): Colon: adjusted Rate Ratio = 1.23 (95% CI 0.61–2.20) Rectum: adjusted Rate Ratio = 1.04 (95% CI 0.28–2.67) |
| Lebwohl et al. [212] | 2010 | USA | Retrospective cohort study | 180 | 23 | No significant increased risk of colorectal adenomas: OR = 0.75 (95% CI 0.41–1.34) |
| Landgren et al. [213] | 2011 | USA | Hospital-based retrospective cohort study | NR | 11 colon cancers 9 rectum cancers | No increased risk: Colon: adjusted RR = 0.85 (95% CI 0.47–1.54) Rectum: adjusted RR = 1.29 (95% CI 0.67–2.48) |
| Grainge et al. [27] | 2012 | United Kingdom | Population-based retrospective cohort study | 435 | 6 | No increased risk of colorectal cancer: SIR = 1.17 (95% CI0.43–2.54) |
| Elfstrom et al. [168] | 2012 | Sweden | Population-based retrospective cohort study | 28,989 | First year of follow-up: 49 colon cancers 14 rectum cancers After 1 year: 88 colon cancers 30 rectum cancers | First year of follow-up: Colon: HR = 7.94 (95% CI5.21–12.1) Rectum: HR = 2.57 (95% CI 1.36–4.86) After 1 year of follow-up: Colon: HR = 1.10 (95% CI 0.87–1.39) Rectum: HR = 0.58 (95% CI 0.40–0.85) |
| Pereyra et al. [226] | 2013 | Argentina | Multicenter retrospective case–control study | 118 | 24 polyps 18 adenomas 3 advanced neoplastic lesions | No increased risk compared to controls. Polyps: OR = 1.25 (95% CI 0.71–2.18) Adenomas: OR = 1.39 (95% CI 0.73–2.63) Advanced neoplastic lesions: OR = 1.00 (95% CI 0.26–3.72) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 133 colon cancers 51 rectum cancers | Increased risk of colon cancer: SIR = 1.35 (95% CI 1.13–1.58) No increased risk of rectum cancer: SIR = 0.82 (95% CI 0.61–1.07) |
| Volta et al. [214] | 2014 | Italy | Multicenter retrospective cohort study | 1757 | 6 | Decreased risk of colon carcinoma compared to the general population: SIR = 0.29 (95% CI 0.07–0.45) |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 448 | No increased risk of colorectal cancer: HR = 1.06 (95% CI 0.96–1.18) |
| Esophagus | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 6 | Increased risk of esophageal cancer: SIR = 4.2 (95% CI 1.6–9.2) |
| Green et al. [29] | 2003 | USA | Hospital-based prospective cohort study | 381 | 3 | Significantly increased risk of esophageal cancer: SIR = 12 (95% CI 6.5–21) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 5 | No increased risk (excluding cases occurred within the first year after CeD diagnosis): adjusted Rate Ratio = 2.58 (95% CI 0.84–6.07) |
| Landgren et al. [213] | 2011 | USA | Hospital-based retrospective cohort study | NR | 11 | Significantly increased risk: adjusted RR = 1.86 (95% CI 1.03–3.36) |
| Grainge et al. [27] | 2012 | United Kingdom | Population-based retrospective cohort study | 435 | 3 | No increased risk: SIR = 2.86 (95% CI 0.59–8.37) |
| Elfstrom et al. [168] | 2012 | Sweden | Population-based retrospective cohort study | 28,989 | First year of follow-up: 4 After 1 year of follow-up: 8 | First year of follow-up: HR = 6.17 (95% CI 1.52–25.0) After 1 year of follow-up: HR = 1.21 (95% CI 0.55–2.65) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 22 | No increased risk: SIR = 1.47 (95% CI 0.92–2.23) |
| van Gils et al. [49] | 2018 | Netherlands | Population-based case–control study | 28 CeD patients with esophageal cancer | 28,070 patients with esophageal cancer and without CeD | No increased risk of esophageal adenocarcinoma: RR = 1.5 (95% CI 0.8–2.6) Increased risk of esophageal squamous cell carcinoma: RR = 3.5 (95% CI 2.1–5.8) |
| Stomach | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 6 | No increased risk: SIR = 0.9 (95% CI 0.3–2.0) |
| Viljamaa et al. [11] | 2006 | Finland | Population-based prospective cohort study | 781 | 2 | No increased risk: SIR = 1.2 (95% CI 0.2–4.5) |
| Silano et al. [26] | 2007 | Italy | Hospital-based prospective cohort study | 1968 | 3 | Slightly increased risk: SIR = 3.0 (95% CI 1.3–4.9) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 8 | No increased risk (excluding cases occurred within the first year after CeD diagnosis): adjusted Rate Ratio = 1.83 (95% CI 0.79–3.62) |
| Elfstrom et al. [168] | 2012 | Sweden | Population-based retrospective cohort study | 28,989 | First year of follow-up: 7 After 1 year of follow-up: 24 | First year of follow-up: HR = 1.67 (95% CI 0.66–4.22) After 1 year of follow-up: HR = 1.13 (95% CI 0.72–1.77) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 37 | No increased risk: SIR = 0.90 (95% CI 0.63–1.23) |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 65 | No increased risk: HR = 1.21 (95% CI 0.91–1.61) |
| Pancreas | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 9 | No statistically significant increase in risk: SIR = 1.0 (95% CI 0.9–3.6) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 2 | No increased risk (excluding cases occurred within the first year after CeD diagnosis): adjusted Rate Ratio = 0.57 (95% CI 0.07–2.05) |
| Landgren et al. [213] | 2011 | USA | Hospital-based retrospective cohort study | NR | 13 | Significantly increased risk: aRR = 2.27 (95% CI 1.22–4.23) |
| Elfstrom et al. [168] | 2012 | Sweden | Population-based retrospective cohort study | 28,989 | First year of follow-up: 26 After 1 year of follow-up: 38 | First year of follow-up: HR = 10.7 (95% CI 5.77–19.7) After 1 year of follow-up: HR = 1.40 (95% CI 0.97–2.02) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 45 | Significantly decreased risk: SIR = 0.73 (95% CI 0.53–0.97). The risk was decreased particularly in females (SIR = 0.59, 95% CI 0.36–0.89) |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 152 | Significantly increased risk: HR = 2.30 (95% CI 1.87–2.82). A significant increased risk persists even after excluding the first-year of follow-up: HR = 1.66 (95% CI 1.32–2.10) |
| Liver | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 11 | Increased risk: SIR = 2.7 (95% CI 1.3–4.7) |
| Elfstrom et al. [168] | 2012 | Sweden | Population-based retrospective cohort study | 28,989 | First year of follow-up: 15 After 1 year of follow-up: 39 | First year of follow-up: HR = 6.05 (95% CI 2.96–12.4) After 1 year of follow-up: HR = 1.78 (95% CI 1.22–2.60) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 24 | No increased risk: SIR = 0.98 (0.63–1.45) |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 115 | Significantly increased risk: HR = 1.80 (95% CI 1.44–2.25). A significant increased risk persists even after excluding the first-year of follow-up: HR = 1.61 (95% CI 1.26–2.05) |
| Breast | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 7 | Significantly decreased risk: SIR = 0.3 (95% CI 0.1–0.5) |
| Green et al. [29] | 2003 | USA | Hospital-based prospective cohort study | 381 | 5 | No increased risk: SIR = 1.2 (95% CI 0.2–7.2) |
| Card et al. [24] | 2004 | United Kingdom | Population-based prospective cohort study | 4732 | 5 | No increased risk: Peridiagnostic period: SIR = 1.26 (95% CI 0.15–4.54) Postdiagnostic period: SIR = 0.59 (95% CI 0.12–1.73) |
| West et al. [25] | 2004 | United Kingdom | Population-based cohort study | 4732 | 8 | Significantly decreased risk: adjusted HR = 0.31 (95% CI 0.15–0.63). The association remained significant after 1 year of follow-up (0.24, 95% CI 0.10–0.60) |
| Viljamaa et al. [11] | 2006 | Finland | Population-based prospective cohort study | 781 | 9 | No significant increased risk: SIR = 0.9 (95% CI 0.4–1.7) |
| Silano et al. [26] | 2007 | Italy | Hospital-based prospective cohort study | 1968 | 3 | Significantly decreased risk: SIR = 0.2 (95% CI 0.04–0.62) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 6 | Borderline decreased risk: SIR = 0.48 (95% CI 0.17–1.04) |
| Lohi et al. [32] | 2009 | Finland | Population-based retrospective cohort study | 73 (EMA + subjects) | 1 | No increased risk: RR = 0.71 (95% CI 0.10–5.07) |
| Grainge et al. [27] | 2012 | United Kingdom | Population-based retrospective cohort study | 435 | 5 | No significant increased risk: SIR = 0.71 (95% CI 0.23–1.66) |
| Ludvigsson et al. [215] | 2012 | Sweden | Population-based retrospective cohort study | 17,852 | 151 | Decreased risk: HR = 0.85 (95% CI 0.72–1.01) Excluding the first year of follow-up: HR = 0.82 (95% CI 0.68–0.99) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 239 | Significantly decreased risk: SIR = 0.70 (95% CI 0.62–0.79) |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 383 | Significantly decreased risk: HR = 0.83 (95% CI 0.74–0.92). A significant increased risk persists even after excluding the first-year of follow-up: HR = 0.81 (95% CI 0.72–0.90) |
| Endometrium and Ovary | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 7 | No decreased risk of ovary cancer: SIR = 1.3 (95% CI 0.5–2.7) |
| Ludvigsson et al. [215] | 2012 | Sweden | Population-based retrospective cohort study | 17,852 | 31 endometrium cancers 27 ovary cancers | Significant decreased risk of endometrial cancer: HR = 0.60 (95% CI 0.41–0.86) No significantly decreased risk of ovary cancer: HR = 0.89 (85% CI 0.59–1.34) |
| Prostate | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 14 | No significantly increased risk: SIR = 0.7 (95% CI 0.4–1.2) |
| West et al. [25] | 2004 | United Kingdom | Population-based cohort study | 4732 | 6 | No significantly increased risk: aHR = 1.05 (95% CI 0.42–2.57) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 4 | No significantly increased risk: adjusted ratio = 0.67 (95% CI 0.18–1.73) |
| Ludvigsson et al. [221] | 2012 | Sweden | Population-based retrospective cohort study | 10,995 | 185 | No increased risk: HR = 0.92 (95% CI 0.79–1.08) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 248 | No significantly increased risk: SIR = 0.97 (95% CI 0.85–1.09) |
| Thyroid | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 1 | No increased risk: SIR = 0.6 (95% CI 0.0–3.3) |
| Kent et al. [222] | 2006 | USA | Monocentric retrospective cohort | 606 | 3 | Significantly increased risk of thyroid papillary cancer: SIR = 22.52 (95% CI 14.90–34.04) |
| Volta et al. [223] | 2011 | Italy | Multicenter retrospective cohort study | 1757 | 6 | Increased risk of thyroid papillary cancer, although not statistically significant: SIR = 2.55 (95% CI 0.93–5.55) |
| Ludvigsson et al. [224] | 2013 | Sweden | Population-based retrospective cohort study | 29,074 | 7 | No increased risk of all thyroid cancers: HR = 0.6 (95% CI 0.3–1.3). No increased risk of papillary thyroid cancer. |
| Lung | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 12 | No increased risk: SIR = 1.0 (95% CI 0.5–1.7) |
| Green et al. [29] | 2003 | USA | Hospital-based prospective cohort study | 381 | 3 | No increased risk: SIR = 0.8 (95% CI 0.1–7.2) |
| Card et al. [24] | 2004 | United Kingdom | Population-based prospective cohort study | 4732 | 8 | No increased risk: Peridiagnostic period: SIR = 1.35 (95% CI 0.16–4.88) Postdiagnostic period: SIR = 1.51 (95% CI 0.55–3.29) |
| West et al. [25] | 2004 | United Kingdom | Population-based cohort study | 4732 | 57 | Borderline significant decreased risk: aHR = 0.37 (95% CI 0.13–1.02) |
| Viljamaa et al. [11] | 2006 | Finland | Population-based prospective cohort study | 781 | 2 | No increased risk: SIR = 0.6 (95% CI 0.1–2.1) |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 13 | No increased risk: adjusted Rate Ratio = 1.07 (95% CI 0.57–1.83) |
| Grainge et al. [27] | 2012 | United Kingdom | Population-based retrospective cohort study | 435 | 6 | No increased risk: SIR = 0.78 (95% CI 0.29–1.69) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 86 | Significantly decreased risk: SIR = 0.60 (95% CI 0.48–0.74). This result was confirmed both in males and females |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 196 | Statistically significant decreased risk when the first-year of follow-up is excluded: HR = 0.84 (95% CI 0.71–0.99) |
| Melanoma | ||||||
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 4 | No increased risk: SIR = 0.6 (95% CI 0.2–1.7) |
| Green et al. [29] | 2003 | USA | Hospital-based prospective cohort study | 381 | 5 | Significantly increased risk: SIR = 5.0 (95% CI 2.1–12.0) |
| Lebwohl et al. [225] | 2014 | Sweden | Population-based retrospective cohort study | 29,028 | 78 | No increased risk: aHR = 0.94 (95% CI 0.73–1.20) |
7. The Protective Effect of a Gluten-Free Diet
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ludvigsson, J.F.; Bai, J.C.; Biagi, F.; Card, T.R.; Ciacci, C.; Ciclitira, P.J.; Green, P.H.R.; Hadjivassiliou, M.; Holdoway, A.; van Heel, D.A.; et al. Diagnosis and management of adult coeliac disease: Guidelines from the British Society of Gastroenterology. Gut 2014, 63, 1210–1228. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Corazza, G.R. Coeliac disease. Lancet 2009, 373, 1480–1493. [Google Scholar] [CrossRef]
- Singh, P.; Arora, A.; Strand, T.A.; Leffler, D.A.; Catassi, C.; Green, P.H.; Kelly, C.P.; Ahuja, V.; Makharia, G.K. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 823–836.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingone, F.; Swift, G.L.; Card, T.R.; Sanders, D.S.; Ludvigsson, J.F.; Bai, J.C. Psychological morbidity of celiac disease: A review of the literature. United Eur. Gastroenterol. J. 2015, 3, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Marsilio, I.; Canova, C.; D’Odorico, A.; Ghisa, M.; Zingone, L.; Lorenzon, G.; Savarino, E.V.; Zingone, F. Quality-of-Life Evaluation in Coeliac Patients on a Gluten-Free Diet. Nutrients 2020, 12, 2981. [Google Scholar] [CrossRef] [PubMed]
- Biagi, F.; Gobbi, P.; Marchese, A.; Borsotti, E.; Zingone, F.; Ciacci, C.; Volta, U.; Caio, G.; Carroccio, A.; Ambrosiano, G.; et al. Low incidence but poor prognosis of complicated coeliac disease: A retrospective multicentre study. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2014, 46, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.J.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef]
- Corrao, G.; Corazza, G.R.; Bagnardi, V.; Brusco, G.; Ciacci, C.; Cottone, M.; Sategna Guidetti, C.; Usai, P.; Cesari, P.; Pelli, M.A.; et al. Mortality in patients with coeliac disease and their relatives: A cohort study. Lancet 2001, 358, 356–361. [Google Scholar] [CrossRef]
- Peters, U.; Askling, J.; Gridley, G.; Ekbom, A.; Linet, M. Causes of death in patients with celiac disease in a population-based Swedish cohort. Arch. Intern. Med. 2003, 163, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Grainge, M.J.; West, J.; Card, T.R.; Holmes, G.K.T. Causes of death in people with celiac disease spanning the pre- and post-serology era: A population-based cohort study from derby, uk. Am. J. Gastroenterol. 2011, 106, 933–939. [Google Scholar] [CrossRef]
- Viljamaa, M.; Kaukinen, K.; Pukkala, E.; Hervonen, K.; Reunala, T.; Collin, P. Malignancies and mortality in patients with coeliac disease and dermatitis herpetiformis: 30-year population-based study. Dig. Liver Dis. 2006, 38, 374–380. [Google Scholar] [CrossRef]
- Tio, M.; Cox, M.R.; Eslick, G.D. Meta-analysis: Coeliac disease and the risk of all-cause mortality, any malignancy and lymphoid malignancy. Aliment. Pharmacol. Ther. 2012, 35, 540–551. [Google Scholar] [CrossRef]
- Solaymani-Dodaran, M.; West, J.; Logan, R.F.A. Long-term mortality in people with celiac disease diagnosed in childhood compared with adulthood: A population-based cohort study. Am. J. Gastroenterol. 2007, 102, 864–870. [Google Scholar] [CrossRef]
- Gough, K.R.; Read, A.; Naish, J. Intestinal reticulosis as a complication of idiopathic steatorrhoea. Gut 1962, 3, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Austad, W.I.; Cornes, J.S.; Gough, K.R.; McCarthy, C.F.; Read, A.E. Steatorrhea and malignant lymphoma. The relationship of malignant tumors of lymphoid tissue and celiac disease. Am. J. Dig. Dis. 1967, 12, 475–490. [Google Scholar] [CrossRef]
- Harris, O.D.; Cooke, W.T.; Thompson, H.; Waterhouse, J.A. Malignancy in adult coeliac disease and idiopathic steatorrhoea. Am. J. Med. 1967, 42, 899–912. [Google Scholar] [CrossRef]
- Swinson, C.M.; Coles, E.C.; Slavin, G.; Booth, C.C. Coeliac disease and malignancy. Lancet 1983, 321, 111–115. [Google Scholar] [CrossRef]
- Cooper, B.T.; Holmes, G.K.; Ferguson, R.; Cooke, W.T. Celiac disease and malignancy. Medicine 1980, 59, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Selby, W.S.; Gallagher, N.D. Malignancy in a 19-year experience of adult celiac disease. Dig. Dis. Sci. 1979, 24, 684–688. [Google Scholar] [CrossRef]
- Nielsen, O.H.; Jacobsen, O.; Pedersen, E.R.; Rasmussen, S.N.; Petri, M.; Laulund, S.; Jarnum, S. Non-tropical sprue. Malignant diseases and mortality rate. Scand. J. Gastroenterol. 1985, 20, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Holmes, G.K.; Prior, P.; Lane, M.R.; Pope, D.; Allan, R.N. Malignancy in coeliac disease--effect of a gluten free diet. Gut 1989, 30, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Collin, P.; Reunala, T.; Pukkala, E.; Laippala, P.; Keyriläinen, O.; Pasternack, A. Coeliac disease—Associated disorders and survival. Gut 1994, 35, 1215–1218. [Google Scholar] [CrossRef]
- Askling, J.; Linet, M.; Gridley, G.; Halstensen, T.S.; Ekström, K.; Ekbom, A. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology 2002, 123, 1428–1435. [Google Scholar] [CrossRef]
- Card, T.R.; West, J.; Holmes, G.K.T. Risk of malignancy in diagnosed coeliac disease: A 24-year prospective, population-based, cohort study. Aliment. Pharmacol. Ther. 2004, 20, 769–775. [Google Scholar] [CrossRef]
- West, J.; Logan, R.F.A.; Smith, C.J.; Hubbard, R.B.; Card, T.R. Malignancy and mortality in people with coeliac disease: Population based cohort study. Br. Med. J. 2004, 329, 716–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silano, M.; Volta, U.; Mecchia, A.; Dessì, M.; Di Benedetto, R.; De Vincenzi, M.; Gasbarrini, G.; De Vitis, D.; Greco, L.; Auricchio, S.; et al. Delayed diagnosis of coeliac disease increases cancer risk. BMC Gastroenterol. 2007, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainge, M.J.; West, J.; Solaymani-Dodaran, M.; Card, T.R.; Logan, R.F.A. The long-term risk of malignancy following a diagnosis of coeliac disease or dermatitis herpetiformis: A cohort study. Aliment. Pharmacol. Ther. 2012, 35, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, B.; Green, P.H.R.; Emilsson, L.; Mårild, K.; Söderling, J.; Roelstraete, B.; Ludvigsson, J.F. Cancer Risk in 47,241 Individuals With Celiac Disease: A Nationwide Cohort Study. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2021, in press. [Google Scholar] [CrossRef]
- Green, P.H.R.; Fleischauer, A.T.; Bhagat, G.; Goyal, R.; Jabri, B.; Neugut, A.I. Risk of malignancy in patients with celiac disease. Am. J. Med. 2003, 115, 191–195. [Google Scholar] [CrossRef]
- Anderson, L.A.; McMillan, S.A.; Watson, R.G.P.; Monaghan, P.; Gavin, A.T.; Fox, C.; Murray, L.J. Malignancy and mortality in a population-based cohort of patients with coeliac disease or “gluten sensitivity”. World J. Gastroenterol. 2007, 13, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Goldacre, M.J.; Wotton, C.J.; Yeates, D.; Seagroatt, V.; Jewell, D. Cancer in patients with ulcerative colitis, Crohn’s disease and coeliac disease: Record linkage study. Eur. J. Gastroenterol. Hepatol. 2008, 20, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Lohi, S.; Mäki, M.; Montonen, J.; Knekt, P.; Pukkala, E.; Reunanen, A.; Kaukinen, K. Malignancies in cases with screening-identified evidence of coeliac disease: A long-term population-based cohort study. Gut 2009, 58, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, W.; Li, P.; Ye, J. Association Between Coeliac Disease and Risk of Any Malignancy and Gastrointestinal Malignancy: A Meta-Analysis. Medicine 2015, 94, e1612. [Google Scholar] [CrossRef] [PubMed]
- Quarpong, W.; Card, T.R.; West, J.; Solaymani-Dodaran, M.; Logan, R.F.; Grainge, M.J. Mortality in people with coeliac disease: Long-term follow-up from a Scottish cohort. United Eur. Gastroenterol. J. 2019, 7, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Lebwohl, B.; Green, P.H.R.; Söderling, J.; Roelstraete, B.; Ludvigsson, J.F. Association Between Celiac Disease and Mortality Risk in a Swedish Population. JAMA 2020, 323, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Canavan, C.; Logan, R.F.; Khaw, K.T.; West, J. No difference in mortality in undetected coeliac disease compared with the general population: A UK cohort study. Aliment. Pharmacol. Ther. 2011, 34, 1012–1019. [Google Scholar] [CrossRef]
- Lohi, S.; Mäki, M.; Rissanen, H.; Knekt, P.; Reunanen, A.; Kaukinen, K. Prognosis of unrecognized coeliac disease as regards mortality: A population-based cohort study. Ann. Med. 2009, 41, 508–515. [Google Scholar] [CrossRef]
- Godfrey, J.D.; Brantner, T.L.; Brinjikji, W.; Christensen, K.N.; Brogan, D.L.; Van Dyke, C.T.; Lahr, B.D.; Larson, J.J.; Rubio-Tapia, A.; Melton, L.J., III; et al. Morbidity and mortality among older individuals with undiagnosed celiac disease. Gastroenterology 2010, 139, 763–769. [Google Scholar] [CrossRef]
- Abdul Sultan, A.; Crooks, C.J.; Card, T.; Tata, L.J.; Fleming, K.M.; West, J. Causes of death in people with coeliac disease in England compared with the general population: A competing risk analysis. Gut 2015, 64, 1220–1226. [Google Scholar] [CrossRef]
- Koskinen, I.; Virta, L.J.; Huhtala, H.; Ilus, T.; Kaukinen, K.; Collin, P. Overall and Cause-Specific Mortality in Adult Celiac Disease and Dermatitis Herpetiformis Diagnosed in the 21st Century. Am. J. Gastroenterol. 2020, 115, 1117–1124. [Google Scholar] [CrossRef]
- Leonard, J.N.; Tucker, W.F.G.; Fry, J.S.; Coulter, C.A.; Boylston, A.W.; McMinn, R.M.; Haffenden, G.P.; Swain, A.F. Increased incidence of malignancy in dermatitis herpetiformis. Br. Med. J. 1983, 286, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, A.; Catassi, C. Current approaches to diagnosis and treatment of celiac disease: An evolving spectrum. Gastroenterology 2001, 120, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Fabiani, E.; Corrao, G.; Barbato, M.; De Renzo, A.; Carella, A.M.; Gabrielli, A.; Leoni, P.; Carroccio, A.; Baldassarre, M.; et al. Risk of non-Hodgkin lymphoma in celiac disease. JAMA 2002, 287, 1413–1419. [Google Scholar] [CrossRef]
- Smedby, K.E.; Åkerman, M.; Hildebrand, H.; Glimelius, B.; Ekbom, A.; Askling, J. Malignant lymphomas in coeliac disease: Evidance of increased risks for lymphoma types other than enteropathy-type T cell lymphoma. Gut 2005, 54, 54–59. [Google Scholar] [CrossRef]
- Elfström, P.; Granath, F.; Ekström Smedby, K.; Montgomery, S.M.; Askling, J.; Ekbom, A.; Ludvigsson, J.F. Risk of lymphoproliferative malignancy in relation to small intestinal histopathology among patients with celiac disease. J. Natl. Cancer Inst. 2011, 103, 436–444. [Google Scholar] [CrossRef]
- Gao, Y.; Kristinsson, S.Y.; Goldin, L.R.; Björkholm, M.; Caporaso, N.E.; Landgren, O. Increased risk for non-Hodgkin lymphoma in individuals with celiac disease and a potential familial association. Gastroenterology 2009, 136, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Smedby, K.E.; Hjalgrim, H.; Askling, J.; Chang, E.T.; Gregersen, H.; Porwit-MacDonald, A.; Sundström, C.; Akerman, M.; Melbye, M.; Glimelius, B.; et al. Autoimmune and chronic inflammatory disorders and risk of non-Hodgkin lymphoma by subtype. J. Natl. Cancer Inst. 2006, 98, 51–60. [Google Scholar] [CrossRef]
- Luisa Mearin, M.; Catassi, C.; Brousse, N.; Brand, R.; Collin, P.; Fabiani, E.; Schweizer, J.J.; Abuzakouk, M.; Szajewska, H.; Hallert, C.; et al. European multi-centre study on coeliac disease and non-Hodgkin lymphoma. Eur. J. Gastroenterol. Hepatol. 2006, 18, 187–194. [Google Scholar] [CrossRef]
- van Gils, T.; Nijeboer, P.; Overbeek, L.I.; Hauptmann, M.; Castelijn, D.A.; Bouma, G.; Mulder, C.J.; van Leeuwen, F.E.; de Jong, D. Risks for lymphoma and gastrointestinal carcinoma in patients with newly diagnosed adult-onset celiac disease: Consequences for follow-up: Celiac disease, lymphoma and GI carcinoma. United Eur. Gastroenterol. J. 2018, 6, 1485–1495. [Google Scholar] [CrossRef] [Green Version]
- Cottone, M.; Termini, A.; Oliva, L.; Magliocco, A.; Marrone, C.; Orlando, A.; Pinzone, F.; Di Mitri, R.; Rosselli, M.; Rizzo, A.; et al. Mortality and causes of death in celiac disease in a Mediterranean area. Dig. Dis. Sci. 1999, 44, 2538–2541. [Google Scholar] [CrossRef] [PubMed]
- Green, P.H.R.; Stavropoulos, S.N.; Panagi, S.G.; Goldstein, S.L.; McMahon, D.J.; Absan, H.; Neugut, A.I. Characteristics of adult celiac disease in the USA: Results of a national survey. Am. J. Gastroenterol. 2001, 96, 126–131. [Google Scholar] [CrossRef]
- Howdle, P.D.; Jalal, P.K.; Holmes, G.K.T.; Houlston, R.S. Primary small-bowel malignancy in the UK and its association with coeliac disease. QJM-Mon. J. Assoc. Phys. 2003, 96, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Farré, C.; Domingo-Domenech, E.; Font, R.; Marques, T.; Fernandez de Sevilla, A.; Alvaro, T.; Villanueva, M.G.; Romagosa, V.; de Sanjose, S. Celiac disease and lymphoma risk: A multicentric case--control study in Spain. Dig. Dis. Sci. 2004, 49, 408–412. [Google Scholar] [CrossRef]
- Anderson, L.A.; Gadalla, S.; Morton, L.M.; Landgren, O.; Pfeiffer, R.; Warren, J.L.; Berndt, S.I.; Ricker, W.; Parsons, R.; Engels, E.A. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int. J. Cancer 2009, 125, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebwohl, B.; Granath, F.; Ekbom, A.; Smedby, K.E.; Murray, J.A.; Neugut, A.I.; Green, P.H.R.; Ludvigsson, J.F. Mucosal healing and risk for lymphoproliferative malignancy in celiac disease: A population-based cohort study. Ann. Intern. Med. 2013, 159, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilus, T.; Kaukinen, K.; Virta, L.J.; Pukkala, E.; Collin, P. Incidence of malignancies in diagnosed celiac patients: A population-based estimate. Am. J. Gastroenterol. 2014, 109, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Fairley, N.H.; Mackie, F.P. The clinical and biochemical syndrome in lymph-adenoma and allied diseases involving the mesenteric lymph glands. Br. Med. J. 1937, 1, 375–381. [Google Scholar] [CrossRef] [Green Version]
- O’Farrelly, C.; Feighery, C.; O’Briain, D.S.; Stevens, F.; Connolly, C.E.; McCarthy, C.; Weir, D.G. Humoral response to wheat protein in patients with coeliac disease and enteropathy associated T cell lymphoma. Br. Med. J. (Clin. Res. Ed.) 1986, 293, 908–910. [Google Scholar] [CrossRef] [Green Version]
- Sharaiha, R.Z.; Lebwohl, B.; Reimers, L.; Bhagat, G.; Green, P.H.; Neugut, A.I. Increasing incidence of enteropathy-associated T-cell lymphoma in the United States, 1973–2008. Cancer 2012, 118, 3786–3792. [Google Scholar] [CrossRef]
- Verbeek, W.H.M.; Van De Water, J.M.W.; Al-Toma, A.; Oudejans, J.J.; Mulder, C.J.J.; Coupé, V.M.H. Incidence of enteropathy - Associated T-cell lymphoma: A nation-wide study of a population-based registry in the Netherlands. Scand. J. Gastroenterol. 2008, 43, 1322–1328. [Google Scholar] [CrossRef]
- Catassi, C.; Bearzi, I.; Holmes, G.K.T. Association of celiac disease and intestinal lymphomas and other cancers. Gastroenterology 2005, 128, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Kryszak, D.; Bhatti, B.; Sturgeon, C.; Helzlsouer, K.; Clipp, S.L.; Gelfond, D.; Puppa, E.; Sferruzza, A.; Fasano, A. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann. Med. 2010, 42, 530–538. [Google Scholar] [CrossRef]
- Rubio-Tapia, A.; Kyle, R.A.; Kaplan, E.L.; Johnson, D.R.; Page, W.; Erdtmann, F.; Brantner, T.L.; Kim, W.R.; Phelps, T.K.; Lahr, B.D.; et al. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology 2009, 137, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cellier, C.; Delabesse, E.; Helmer, C.; Patey, N.; Matuchansky, C.; Jabri, B.; MacIntyre, E.; Cerf-Bensussan, N.; Brousse, N. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. Lancet 2000, 356, 203–208. [Google Scholar] [CrossRef]
- Rubio-Tapia, A.; Murray, J.A. Classification and management of refractory coeliac disease. Gut 2010, 59, 547–557. [Google Scholar] [CrossRef]
- Malamut, G.; Afchain, P.; Verkarre, V.; Lecomte, T.; Amiot, A.; Damotte, D.; Bouhnik, Y.; Colombel, J.F.; Delchier, J.C.; Allez, M.; et al. Presentation and Long-Term Follow-up of Refractory Celiac Disease: Comparison of Type I With Type II. Gastroenterology 2009, 136, 81–90. [Google Scholar] [CrossRef]
- Al-toma, A.; Verbeek, W.H.M.; Hadithi, M.; Von Blomberg, B.M.E.; Mulder, C.J.J. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: Retrospective evaluation of single-centre experience. Gut 2007, 56, 1373–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daum, S.; Ipczynski, R.; Schumann, M.; Wahnschaffe, U.; Zeitz, M.; Ullrich, R. High rates of complications and substantial mortality in both types of refractory sprue. Eur. J. Gastroenterol. Hepatol. 2009, 21, 66–70. [Google Scholar] [CrossRef]
- van Wanrooij, R.L.J.; Bouma, G.; Bontkes, H.J.; Neefjes-Borst, A.; van Grieken, N.C.; von Blomberg, B.M.E.; Mulder, C.J.J. Outcome of Referrals for Non-Responsive Celiac Disease in a Tertiary Center: Low Incidence of Refractory Celiac Disease in the Netherlands. Clin. Transl. Gastroenterol. 2017, 8, e218. [Google Scholar] [CrossRef]
- Biagi, F.; Poggioli, G.; Mazzoni, G.; Corazza, G.R. Intestinal strictures. Lancet 1998, 352, 876. [Google Scholar] [CrossRef]
- Biagi, F.; Lorenzini, P.; Corazza, G.R. Literature review on the clinical relationship between ulcerative jejunoileitis, coeliac disease, and enteropathy-associated T-cell. Scand. J. Gastroenterol. 2000, 35, 785–790. [Google Scholar] [CrossRef]
- Mention, J.J.; Ahmed, M.B.; Bègue, B.; Barbe, U.; Verkarre, V.; Asnafi, V.; Colombel, J.F.; Cugnenc, P.H.; Ruemmele, F.M.; McIntyre, E.; et al. Interleukin 15: A key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology 2003, 125, 730–745. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Ciccocioppo, R.; Cupelli, F.; Cinque, B.; Millimaggi, D.; Clarkson, M.M.; Paulli, M.; Cifone, M.G.; Corazza, G.R. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 2006, 55, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Malamut, G.; El Machhour, R.; Montcuquet, N.; Martin-Lannerée, S.; Dusanter-Fourt, I.; Verkarre, V.; Mention, J.J.; Rahmi, G.; Kiyono, H.; Butz, E.A.; et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J. Clin. Investig. 2010, 120, 2131–2143. [Google Scholar] [CrossRef] [Green Version]
- Malamut, G.; Chandesris, O.; Verkarre, V.; Meresse, B.; Callens, C.; Macintyre, E.; Bouhnik, Y.; Gornet, J.M.; Allez, M.; Jian, R.; et al. Enteropathy associated T cell lymphoma in celiac disease: A large retrospective study. Dig. Liver Dis. 2013, 45, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Ripoche, J.; Heyman, M.; Cerf-Bensussan, N. Celiac disease: From oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009, 2, 8–23. [Google Scholar] [CrossRef]
- Perfetti, V.; Baldanti, F.; Lenti, M.V.; Vanoli, A.; Biagi, F.; Gatti, M.; Riboni, R.; Dallera, E.; Paulli, M.; Pedrazzoli, P.; et al. Detection of Active Epstein-Barr Virus Infection in Duodenal Mucosa of Patients With Refractory Celiac Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016, 14, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, P.C.; Jiwa, N.M.; Oudejans, J.J.; Radaszkiewicz, T.; Meijer, C.J. Epstein-Barr virus in primary gastrointestinal T cell lymphomas. Association with gluten-sensitive enteropathy, pathological features, and immunophenotype. Am. J. Pathol. 1995, 146, 861–867. [Google Scholar]
- Ilyas, M.; Niedobitek, G.; Agathanggelou, A.; Barry, R.E.; Read, A.E.; Tierney, R.; Young, L.S.; Rooney, N. Non-Hodgkin’s lymphoma, coeliac disease, and Epstein-Barr virus: A study of 13 cases of enteropathy-associated T- and B-cell lymphoma. J. Pathol. 1995, 177, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.V.; Egan, L.J.; Connolly, C.E.; Stevens, F.M.; Egan, E.L.; McCarthy, C.F. Enteropathy-associated T-cell lymphoma in the West of Ireland: Low-frequency of Epstein-Barr virus in these tumors. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 1995, 8, 753–757. [Google Scholar]
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; ISBN 9789283244943. [Google Scholar]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delabie, J.; Holte, H.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Savage, K.J.; Connors, J.M.; Rimsza, L.; Harris, N.L.; Müller-Hermelink, K.; et al. Enteropathy-associated T-cell lymphoma: Clinical and histological findings from the international peripheral T-Cell lymphoma project. Blood 2011, 118, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Reddy, V.; Patel, C.R.; Dhall, D.; Lee, G.; Meng-Jun, X.; Al Diffalha, S. Monomorphic Epitheliotropic Intestinal T-cell Lymphoma: A Study of Four Cases and Review of Literature. Ann. Clin. Lab. Sci. 2020, 50, 806–812. [Google Scholar] [PubMed]
- Chott, A.; Haedicke, W.; Mosberger, I.; Fodinger, M.; Winkler, K.; Mannhalter, C.; Muller-Hermelink, H.K. Most CD56+ intestinal lymphomas are CD8+CD5- T-cell lymphomas of monomorphic small to medium size histology. Am. J. Pathol. 1998, 153, 1483–1490. [Google Scholar] [CrossRef]
- deLeeuw, R.J.; Zettl, A.; Klinker, E.; Haralambieva, E.; Trottier, M.; Chari, R.; Ge, Y.; Gascoyne, R.D.; Chott, A.; Müller-Hermelink, H.K.; et al. Whole-Genome Analysis and HLA Genotyping of Enteropathy-Type T-Cell Lymphoma Reveals 2 Distinct Lymphoma Subtypes. Gastroenterology 2007, 132, 1902–1911. [Google Scholar] [CrossRef]
- Lenti, M.V.; Biagi, F.; Lucioni, M.; Di Sabatino, A.; Paulli, M.; Corazza, G.R. Two cases of monomorphic epitheliotropic intestinal T-cell lymphoma associated with coeliac disease. Scand. J. Gastroenterol. 2019, 54, 965–968. [Google Scholar] [CrossRef]
- Tse, E.; Gill, H.; Loong, F.; Kim, S.J.; Ng, S.B.; Tang, T.; Ko, Y.H.; Chng, W.J.; Lim, S.T.; Kim, W.S.; et al. Type II enteropathy-associated T-cell lymphoma: A multicenter analysis from the Asia Lymphoma Study Group. Am. J. Hematol. 2012, 87, 663–668. [Google Scholar] [CrossRef]
- Gale, J.; Simmonds, P.D.; Mead, G.M.; Sweetenham, J.W.; Wright, D.H. Enteropathy-type intestinal T-cell lymphoma: Clinical features and treatment of 31 patients in a single center. J. Clin. Oncol. 2000, 18, 795–803. [Google Scholar] [CrossRef]
- Daum, S.; Ullrich, R.; Heise, W.; Dederke, B.; Foss, H.D.; Stein, H.; Thiel, E.; Zeitz, M.; Riecken, E.O. Intestinal non-Hodgkin’s lymphoma: A multicenter prospective clinical study from the German Study Group on intestinal non-Hodgkin’s lymphoma. J. Clin. Oncol. 2003, 21, 2740–2746. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Biagi, F.; Gobbi, P.G.; Corazza, G.R. How I treat enteropathy-associated T-cell lymphoma. Blood 2012, 119, 2458–2468. [Google Scholar] [CrossRef] [Green Version]
- Wierdsma, N.J.; Nijeboer, P.; de van der Schueren, M.A.E.; Berkenpas, M.; van Bodegraven, A.A.; Mulder, C.J.J. Refractory celiac disease and EATL patients show severe malnutrition and malabsorption at diagnosis. Clin. Nutr. 2016, 35, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.J.; Novakovic, S.; Frkovic-Grazio, S. A single-center report on clinical features and treatment response in patients with intestinal T cell non-Hodgkin’s lymphomas. Oncol. Rep. 2006, 16, 191–195. [Google Scholar] [PubMed]
- Domizio, P.; Owen, R.A.; Shepherd, N.A.; Talbot, I.C.; Norton, A.J. Primary lymphoma of the small intestine: A clinicopathological study of 119 cases. Am. J. Surg. Pathol. 1993, 17, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Hovenga, S.; de Graaf, H.; Joosten, P.; van den Berg, G.A.; Storm, H.; Langerak, A.W.; Kluin, P.M.; Kibbelaar, R.E. Enteropathy-associated T-cell lymphoma presenting with eosinophilia. Neth. J. Med. 2003, 61, 25–27. [Google Scholar] [PubMed]
- McBride, O.M.B.; Skipworth, R.J.E.; Leitch, D.; Yalamarthi, S. Cavitating mesenteric lymph node syndrome in association with coeliac disease and enteropathy associated T-cell lymphoma: A case report and review of the literature. Case Rep. Med. 2010, 2010, 1–4. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Rosado, M.M.; Cazzola, P.; Riboni, R.; Biagi, F.; Carsetti, R.; Corazza, G.R. Splenic hypofunction and the spectrum of autoimmune and malignant complications in celiac disease. Clin. Gastroenterol. Hepatol. 2006, 4, 179–186. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Carsetti, R.; Corazza, G.R. Post-splenectomy and hyposplenic states. Lancet 2011, 378, 86–97. [Google Scholar] [CrossRef]
- International Non-Hodgkin’s Lymphoma Prognostic Factors Project. A Predictive Model for Aggressive Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 1993, 329, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Rohatiner, A.; D’Amore, F.; Coiffier, B.; Crowther, D.; Gospodarowicz, M.; Isaacson, P.; Lister, T.A.; Norton, A.; Salem, P.; Shipp, M. Report on a workshop convened to discuss the pathological and staging classifications of gastrointestinal tract lymphoma. Ann. Oncol. 1994, 5, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Bishton, M.J.; Haynes, A.P. Combination chemotherapy followed by autologous stem cell transplant for enteropathy-associated T cell lymphoma. Br. J. Haematol. 2007, 136, 111–113. [Google Scholar] [CrossRef]
- Vose, J.M.; Neumann, M.; Harris, M.E. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes international T-cell lymphoma project. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Savage, K.J.; Chhanabhai, M.; Gascoyne, R.D.; Connors, J.M. Characterization of peripheral T-cell lymphomas in a single North American institution by the WHO classification. Ann. Oncol. 2004, 15, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Gallamini, A.; Stelitano, C.; Calvi, R.; Bellei, M.; Mattei, D.; Vitolo, U.; Morabito, F.; Martelli, M.; Brusamolino, E.; Iannitto, E.; et al. Peripheral T-cell lymphoma unspecified (PTCL-U): A new prognostic model from a retrospective multicentric clinical study. Blood 2004, 103, 2474–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Baaij, L.R.; Berkhof, J.; Van De Water, J.M.W.; Sieniawski, M.K.; Radersma, M.; Verbeek, W.H.M.; Visser, O.J.; Oudejans, J.J.; Meijer, C.J.L.M.; Mulder, C.J.J.; et al. A new and validated clinical prognostic model (EPI) for enteropathy-associated T-cell lymphoma. Clin. Cancer Res. 2015, 21, 3013–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leighton, J.A. The role of endoscopic imaging of the small bowel in clinical practice. Am. J. Gastroenterol. 2011, 106, 27–36. [Google Scholar] [CrossRef]
- Al-Toma, A.; Verbeek, W.H.M.; Mulder, C.J.J. The management of complicated celiac disease. Dig. Dis. 2007, 25, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Mallant, M.; Hadithi, M.; Al-Toma, A.B.; Kater, M.; Jacobs, M.; Manoliu, R.; Mulder, C.; van Waesberghe, J.H.T.M. Abdominal computed tomography in refractory coeliac disease and enteropathy associated T-cell lymphoma. World J. Gastroenterol. 2007, 13, 1696–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadithi, M.; Mallant, M.; Oudejans, J.; van Waesberghe, J.-H.T.M.; Mulder, C.J.; Comans, E.F.I. 18F-FDG PET versus CT for the detection of enteropathy-associated T-cell lymphoma in refractory celiac disease. J. Nucl. Med. 2006, 47, 1622–1627. [Google Scholar] [PubMed]
- Hoffmann, M.; Vogelsang, H.; Kletter, K.; Zettinig, G.; Chott, A.; Raderer, M. 18F-fluoro-deoxy-glucose positron emission tomography (18F-FDG-PET) for assessment of enteropathy-type T cell lymphoma. Gut 2003, 52, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Laird, J.; Leach, M.; Ballantyne, S. The value of small bowel magnetic resonance imaging in the management of enteropathy associated T-cell lymphoma. Br. J. Haematol. 2008, 142, 136–137. [Google Scholar] [CrossRef]
- Van Weyenberg, S.J.B.; Meijerink, M.R.; Jacobs, M.A.J.M.; Van Kuijk, C.; Mulder, C.J.; Van Waesberghe, J.H.T.M. MR enteroclysis in refractory celiac disease: Proposal and validation of a severity scoring system. Radiology 2011, 259, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elli, L.; Casazza, G.; Locatelli, M.; Branchi, F.; Ferretti, F.; Conte, D.; Fraquelli, M. Use of enteroscopy for the detection of malignant and premalignant lesions of the small bowel in complicated celiac disease: A meta-analysis. Gastrointest. Endosc. 2017, 86, 264–273.e1. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Wanschaffe, U.; Glasenapp, R.; Borchert, M.; Ullrich, R.; Zeitz, M.; Faiss, S. Capsule endoscopy in refractory celiac disease. Endoscopy 2007, 39, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, F.; Branchi, F.; Orlando, S.; Roncoroni, L.; Barigelletti, G.; Fabiano, S.; Vecchi, M.; Penagini, R.; Doneda, L.; Elli, L. Effectiveness of Capsule Endoscopy and Double-Balloon Enteroscopy in Suspected Complicated Celiac Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, in press. [Google Scholar] [CrossRef]
- Hadithi, M.; Al-Toma, A.; Oudejans, J.; Van Bodegraven, A.A.; Mulder, C.J.; Jacobs, M. The value of double-balloon enteroscopy in patients with refractory celiac disease. Am. J. Gastroenterol. 2007, 102, 987–996. [Google Scholar] [CrossRef]
- Daum, S.; Foss, H.-D.; Anagnostopoulos, I.; Dederke, B.; Demel, G.; Araujo, I.; Riecken, E.-O.; Stein, H. Expression of cytotoxic molecules in intestinal T-cell lymphomas. J. Pathol. 1997, 182, 311–317. [Google Scholar] [CrossRef]
- De Bruin, P.C.; Connolly, C.E.; Oudejans, J.J.; Kummer, J.A.; Jansen, W.; Mccarthy, C.F.; Meijer, C.J.L.M. Enteropathy-associated T-cell lymphomas have a cytotoxic T-cell phenotype. Histopathology 1997, 31, 313–317. [Google Scholar] [CrossRef]
- Daum, S.; Weiss, D.; Hummel, M.; Ullrich, R.; Heise, W.; Stein, H.; Riecken, E.O.; Foss, H.D. Frequency of clonal intraepithelial T lymphocyte proliferations in enteropathy-type intestinal T cell lymphoma, coeliac disease, and refractory sprue. Gut 2001, 49, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Verkarre, V.; Asnafi, V.; Lecomte, T.; Patey Mariaud-de Serre, N.; Leborgne, M.; Grosdidier, E.; Le Bihan, C.; Macintyre, E.; Cellier, C.; Cerf-Bensussan, N.; et al. Refractory coeliac sprue is a diffuse gastrointestinal disease. Gut 2003, 52, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Verbeek, W.H.M.; Goerres, M.S.; von Blomberg, B.M.E.; Oudejans, J.J.; Scholten, P.E.T.; Hadithi, M.; Al-Toma, A.; Schreurs, M.W.J.; Mulder, C.J.J. Flow cytometric determination of aberrant intra-epithelial lymphocytes predicts T-cell lymphoma development more accurately than T-cell clonality analysis in Refractory Celiac Disease. Clin. Immunol. 2008, 126, 48–56. [Google Scholar] [CrossRef]
- Hussein, S.; Gindin, T.; Lagana, S.M.; Arguelles-Grande, C.; Krishnareddy, S.; Alobeid, B.; Lewis, S.K.; Mansukhani, M.M.; Green, P.H.R.; Bhagat, G. Clonal T cell receptor gene rearrangements in coeliac disease: Implications for diagnosing refractory coeliac disease. J. Clin. Pathol. 2018, 71, 825–831. [Google Scholar] [CrossRef]
- Celli, R.; Hui, P.; Triscott, H.; Bogardus, S.; Gibson, J.; Hwang, M.; Robert, M.E. Clinical Insignficance of Monoclonal T-Cell Populations and Duodenal Intraepithelial T-Cell Phenotypes in Celiac and Nonceliac Patients. Am. J. Surg. Pathol. 2019, 43, 151–160. [Google Scholar] [CrossRef]
- Patey-Mariaud De Serre, N.; Cellier, C.; Jabri, B.; Delabesse, E.; Verkarre, V.; Roche, B.; Lavergne, A.; Brière, J.; Mauvieux, L.; Leborgne, M.; et al. Distinction between coeliac disease and refractory sprue: A simple immunohistochemical method. Histopathology 2000, 37, 70–77. [Google Scholar] [CrossRef]
- Rubio-Tapia, A.; Kelly, D.G.; Lahr, B.D.; Dogan, A.; Wu, T.T.; Murray, J.A. Clinical Staging and Survival in Refractory Celiac Disease: A Single Center Experience. Gastroenterology 2009, 136, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Brais, R.; Lavergne-Slove, A.; Jeng, Q.; Payne, K.; Ye, H.; Liu, Z.; Carreras, J.; Huang, Y.; Bacon, C.M.; et al. Continual monitoring of intraepithelial lymphocyte immunophenotype and clonality is more important than snapshot analysis in the surveillance of refractory coeliac disease. Gut 2010, 59, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Al-Toma, A.; Goerres, M.S.; Meijer, J.W.R.; Peña, A.S.; Crusius, J.B.A.; Mulder, C.J.J. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin. Gastroenterol. Hepatol. 2006, 4, 315–319. [Google Scholar] [CrossRef]
- Wolters, V.M.; Verbeek, W.H.M.; Zhernakova, A.; Onland-Moret, C.; Schreurs, M.W.J.; Monsuur, A.J.; Verduijn, W.; Wijmenga, C.; Mulder, C.J.J. The MYO9B Gene Is a Strong Risk Factor for Developing Refractory Celiac Disease. Clin. Gastroenterol. Hepatol. 2007, 5, 1399–1405.e2. [Google Scholar] [CrossRef]
- Zettl, A.; Ott, G.; Makulik, A.; Katzenberger, T.; Starostik, P.; Eichler, T.; Puppe, B.; Bentz, M.; Müller-Hermelink, H.K.; Chott, A. Chromosomal gains at 9q characterize enteropathy-type T-cell lymphoma. Am. J. Pathol. 2002, 161, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Baumgärtner, A.K.; Zettl, A.; Chott, A.; Ott, G.; Müller-Hermelink, H.K.; Starostik, P. High Frequency of Genetic Aberrations in Enteropathy-Type T-Cell Lymphoma. Lab. Investig. 2003, 83, 1509–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cording, S.; Lhermitte, L.; Malamut, G.; Berrabah, S.; Trinquand, A.; Guegan, N.; Villarese, P.; Kaltenbach, S.; Meresse, B.; Khater, S.; et al. Oncogenetic landscape of lymphomagenesis in coeliac disease. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Roshan, B.; Leffler, D.A.; Jamma, S.; Dennis, M.; Sheth, S.; Falchuk, K.; Najarian, R.; Goldsmith, J.; Tariq, S.; Schuppan, D.; et al. The incidence and clinical spectrum of refractory celiac disease in a north american referral center. Am. J. Gastroenterol. 2011, 106, 923–928. [Google Scholar] [CrossRef]
- Raderer, M.; Troch, M.; Kiesewetter, B.; Püspök, A.; Jaeger, U.; Hoffmann, M.; Chott, A. Second line chemotherapy in patients with enteropathy-associated T cell lymphoma: A retrospective single center analysis. Ann. Hematol. 2012, 91, 57–61. [Google Scholar] [CrossRef]
- Hönemann, D.; Prince, H.M.; Hicks, R.J.; Seymour, J.F. Enteropathy-associated T-cell lymphoma without a prior diagnosis of coeliac disease: Diagnostic dilemmas and management options. Ann. Hematol. 2005, 84, 118–121. [Google Scholar] [CrossRef]
- Egan, L.; Walsh, S.; Stevens, F.; Connolly, C.; Egan, E.; McCarthy, C. Celiac-associated lymphoma. A single institution experience of 30 cases in the combination chemotherapy era. J. Clin. Gastroenterol. 1995, 21, 123–129. [Google Scholar] [CrossRef]
- Wöhrer, S.; Chott, A.; Drach, J.; Püspök, A.; Hejna, M.; Hoffmann, M.; Raderer, M. Chemotherapy with cyclophosphamide, doxorubicin, etoposide, vincristine and prednisone (CHOEP) is not effective in patients with enteropathy-type intestinal T-cell lymphoma. Ann. Oncol. 2004, 15, 1680–1683. [Google Scholar] [CrossRef]
- Sieniawski, M.; Angamuthu, N.; Boyd, K.; Chasty, R.; Davies, J.; Forsyth, P.; Jack, F.; Lyons, S.; Mounter, P.; Revell, P.; et al. Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood 2010, 115, 3664–3670. [Google Scholar] [CrossRef] [Green Version]
- Kluin-Nelemans, H.C.; van Marwijk Kooy, M.; Lugtenburg, P.J.; van Putten, W.L.J.; Luten, M.; Oudejans, J.; van Imhoff, G.W. Intensified alemtuzumab-CHOP therapy for peripheral T-cell lymphoma. Ann. Oncol. 2011, 22, 1595–1600. [Google Scholar] [CrossRef]
- Trumper, L.H.; Wulf, G.; Ziepert, M.; D’Amore, F.; Held, G.; Greil, R.; Tournilhac, O.; Relander, T.; Viardot, A.; Wilhelm, M.; et al. Alemtuzumab added to CHOP for treatment of peripheral T-cell lymphoma (pTNHL) of the elderly: Final results of 116 patients treated in the international ACT-2 phase III trial. J. Clin. Oncol. 2016, 34, 7500. [Google Scholar] [CrossRef]
- Wulf, G.G.; Altmann, B.; Ziepert, M.; D’Amore, F.; Held, G.; Greil, R.; Tournilhac, O.; Relander, T.; Viardot, A.; Wilhelm, M.; et al. Alemtuzumab plus CHOP versus CHOP in elderly patients with peripheral T-cell lymphoma: The DSHNHL2006-1B/ACT-2 trial. Leukemia 2021, 35, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Gallamini, A.; Zaja, F.; Patti, C.; Billio, A.; Specchia, M.R.; Tucci, A.; Levis, A.; Manna, A.; Secondo, V.; Rigacci, L.; et al. Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: Results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood 2007, 110, 2316–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldini, D.; Mora, O.; Cavalli, F.; Zucca, E.; Mazzucchelli, L. Efficacy of alemtuzumab and gemcitabine in a patient with enteropathy-type T-cell lymphoma. Br. J. Haematol. 2008, 142, 484–486. [Google Scholar] [CrossRef]
- Kircher, S.M.; Gurbuxani, S.; Smith, S.M. CHOP Plus Alemtuzumab can Induce Metabolic Response by FDG-PET but has Minimal Long-term benefits: A Case Report and Literature Review. J. Gastrointest. Cancer 2007, 38, 59–62. [Google Scholar] [CrossRef]
- Al-toma, A.; Goerres, M.S.; Meijer, J.W.R.; von Blomberg, B.M.E.; Wahab, P.J.; Kerckhaert, J.A.M.; Mulder, C.J.J. Cladribine Therapy in Refractory Celiac Disease With Aberrant T Cells. Clin. Gastroenterol. Hepatol. 2006, 4, 1322–1327. [Google Scholar] [CrossRef]
- Tack, G.J.; Verbeek, W.H.M.; Al-Toma, A.; Kuik, D.J.; Schreurs, M.W.J.; Visser, O.; Mulder, C.J.J. Evaluation of cladribine treatment in refractory celiac disease type II. World J. Gastroenterol. 2011, 17, 506–513. [Google Scholar] [CrossRef]
- Piekarz, R.L.; Frye, R.; Prince, H.M.; Kirschbaum, M.H.; Zain, J.; Allen, S.L.; Jaffe, E.S.; Ling, A.; Turner, M.; Peer, C.J.; et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 2011, 117, 5827–5834. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, W.F.; Caldwell, M.E.; Reddy, N. Brentuximab in the treatment of CD30-positive enteropathy-associated T-cell lymphoma. J. Natl. Compr. Canc. Netw. 2013, 11, 137–140. [Google Scholar] [CrossRef] [Green Version]
- Nijeboer, P.; Malamut, G.; Mulder, C.J.; Cerf-Bensussan, N.; Sibon, D.; Bouma, G.; Cellier, C.; Hermine, O.; Visser, O. Enteropathy-associated T-cell lymphoma: Improving treatment strategies. Dig. Dis. 2015, 33, 231–235. [Google Scholar] [CrossRef]
- Blystad, A.; Enblad, G.; Kvaløy, S.; Berglund, Å.; Delabie, J.; Holte, H.; Carlson, K.; Kvalheim, G.; Bengtsson, M.; Hagberg, H. High-dose therapy with autologous stem cell transplantation in patients with peripheral T cell lymphomas. Bone Marrow Transplant. 2001, 27, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Nomura, J.; Tateno, H.; Kameoka, J.; Sasaki, T. CD56 Positive Intestinal T-Cell Lymphoma: Treatment with High Dose Chemotherapy and Autologous Peripheral Blood Stem Cell Transplantation. Intern. Med. 2002, 41, 734–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chonabayashi, K.; Kondo, T.; Tanaka, Y.; Ichinohe, T.; Ishikawa, T.; Uchiyama, T. Sustained complete remission of refractory enteropathy-type T-cell lymphoma following reduced-intensity unrelated cord blood transplantation. Bone Marrow Transplant. 2007, 40, 905–906. [Google Scholar] [CrossRef] [PubMed]
- Jantunen, E.; Juvonen, E.; Wiklund, T.; Putkonen, M.; Nousiainen, T. High-dose Therapy Supported by Autologous Stem Cell Transplantation in Patients with Enteropathy-associated T-cell Lymphoma. Leuk. Lymphoma 2003, 44, 2163–2164. [Google Scholar] [CrossRef]
- Rongey, C.; Micallef, I.; Smyrk, T.; Murray, J. Successful Treatment of Enteropathy-Associated T Cell Lymphoma with Autologous Stem Cell Transplant. Dig. Dis. Sci. 2006, 51, 1082–1086. [Google Scholar] [CrossRef]
- Al-toma, A.; Verbeek, W.H.M.; Visser, O.J.; Kuijpers, K.C.; Oudejans, J.J.; Kluin-Nelemans, H.C.; Mulder, C.J.J.; Huijgens, P.C. Disappointing outcome of autologous stem cell transplantation for enteropathy-associated T-cell lymphoma. Dig. Liver Dis. 2007, 39, 634–641. [Google Scholar] [CrossRef]
- Nava, V.E.; Cohen, P.; Bishop, M.; Fowler, D.; Jaffe, E.S.; Ozdemirli, M. Enteropathy-type T-cell Lymphoma After Intestinal Diffuse Large B-cell Lymphoma. Am. J. Surg. Pathol. 2007, 31, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Reimer, P.; Rüdiger, T.; Geissinger, E.; Weissinger, F.; Nerl, C.; Schmitz, N.; Engert, A.; Einsele, H.; Müller-Hermelink, H.K.; Wilhelm, M. Autologous Stem-Cell Transplantation As First-Line Therapy in Peripheral T-Cell Lymphomas: Results of a Prospective Multicenter Study. J. Clin. Oncol. 2009, 27, 106–113. [Google Scholar] [CrossRef]
- Prochazka, V.; Faber, E.; Raida, L.; Papajik, T.; Vondrakova, J.; Rusinakova, Z.; Kucerova, L.; Myslivecek, M.; Indrak, K. Long-term outcome of patients with peripheral T-cell lymphoma treated with first-line intensive chemotherapy followed by autologous stem cell transplantation. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech Repub. 2011, 155, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Nijeboer, P.; de Baaij, L.R.; Visser, O.; Witte, B.I.; Cillessen, S.A.G.M.; Mulder, C.J.; Bouma, G. Treatment response in enteropathy associated T-cell lymphoma; survival in a large multicenter cohort. Am. J. Hematol. 2015, 90, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Leslie, L.A.; Lebwohl, B.; Neugut, A.I.; Gregory Mears, J.; Bhagat, G.; Green, P.H.R. Incidence of lymphoproliferative disorders in patients with celiac disease. Am. J. Hematol. 2012, 87, 754–759. [Google Scholar] [CrossRef] [PubMed]
- du Pré, M.F.; Blazevski, J.; Dewan, A.E.; Stamnaes, J.; Kanduri, C.; Sandve, G.K.; Johannesen, M.K.; Lindstad, C.B.; Hnida, K.; Fugger, L.; et al. B cell tolerance and antibody production to the celiac disease autoantigen transglutaminase 2. J. Exp. Med. 2020, 217, e20190860. [Google Scholar] [CrossRef]
- Ansell, P.; Simpson, J.; Lightfoot, T.; Smith, A.; Kane, E.; Howell, D.; Newton, R.; McGonagle, D.; Jack, A.; Roman, E. Non-Hodgkin lymphoma and autoimmunity: Does gender matter? Int. J. Cancer 2011, 129, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Rubio-Tapia, A.; Ristow, K.M.; Habermann, T.M.; Murray, J.A.; Inwards, D.J. Patients With Celiac Disease and B-Cell Lymphoma Have a Better Prognosis Than Those With T-Cell Lymphoma. Clin. Gastroenterol. Hepatol. 2010, 8, 1042–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J.F.; Lebwohl, B.; Rubio-Tapia, A.; Murray, J.A.; Green, P.H.R.; Ekbom, A.; Granath, F. Does celiac disease influence survival in lymphoproliferative malignancy? Eur. J. Epidemiol. 2013, 28, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuffrida, P.; Vanoli, A.; Arpa, G.; Bonometti, A.; Luinetti, O.; Solcia, E.; Corazza, G.; Paulli, M.; Di Sabatino, A. Small Bowel Carcinomas Associated with Immune-Mediated Intestinal Disorders: The Current Knowledge. Cancers 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, T.; Zaanan, A.; Svrcek, M.; Laurent-Puig, P.; Carrere, N.; Manfredi, S.; Locher, C.; Afchain, P. Small bowel adenocarcinoma: Epidemiology, risk factors, diagnosis and treatment. Dig. Liver Dis. 2014, 46, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Bilimoria, K.Y.; Bentrem, D.J.; Wayne, J.D.; Ko, C.Y.; Bennett, C.L.; Talamonti, M.S. Small bowel cancer in the United States: Changes in epidemiology, treatment, and survival over the last 20 years. Ann. Surg. 2009, 249, 63–71. [Google Scholar] [CrossRef]
- Faivre, J.; Trama, A.; De Angelis, R.; Elferink, M.; Siesling, S.; Audisio, R.; Bosset, J.F.; Cervantes, A.; Lepage, C.; RARECARE Working Group. Incidence, prevalence and survival of patients with rare epithelial digestive cancers diagnosed in Europe in 1995-2002. Eur. J. Cancer 2012, 48, 1417–1424. [Google Scholar] [CrossRef]
- Elfström, P.; Granath, F.; Ye, W.; Ludvigsson, J.F. Low risk of gastrointestinal cancer among patients with celiac disease, inflammation, or latent celiac disease. Clin. Gastroenterol. Hepatol. 2012, 10, 30–36. [Google Scholar] [CrossRef]
- Emilsson, L.; Semrad, C.; Lebwohl, B.; Green, P.H.R.; Ludvigsson, J.F. Risk of Small Bowel Adenocarcinoma, Adenomas, and Carcinoids in a Nationwide Cohort of Individuals With Celiac Disease. Gastroenterology 2020, 159, 1686–1694.e2. [Google Scholar] [CrossRef]
- Rampertab, S.D.; Forde, K.A.; Green, P.H.R. Small bowel neoplasia in coeliac disease. Gut 2003, 52, 1211–1214. [Google Scholar] [CrossRef] [Green Version]
- Vanoli, A.; Di Sabatino, A.; Furlan, D.; Klersy, C.; Grillo, F.; Fiocca, R.; Mescoli, C.; Rugge, M.; Nesi, G.; Fociani, P.; et al. Small bowel carcinomas in coeliac or Crohn’s disease: Clinico-pathological, molecular, and prognostic features. A study from the small bowel cancer Italian consortium. J. Crohn’s Colitis 2017, 11, 942–953. [Google Scholar] [CrossRef]
- Potter, D.D.; Murray, J.A.; Donohue, J.H.; Burgart, L.J.; Nagorney, D.M.; Van Heerden, J.A.; Plevak, M.F.; Zinsmeister, A.R.; Thibodeau, S.N. The role of defective mismatch repair in small bowel adenocarcinoma in celiac disease. Cancer Res. 2004, 64, 7073–7077. [Google Scholar] [CrossRef] [Green Version]
- Diosdado, B.; Buffart, T.E.; Watkins, R.; Carvalho, B.; Ylstra, B.; Tijssen, M.; Bolijn, A.S.; Lewis, F.; Maude, K.; Verbeke, C.; et al. High-resolution array comparative genomic hybridization in sporadic and celiac disease-related small bowel adenocarcinomas. Clin. Cancer Res. 2010, 16, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Bruno, C.J.; Batts, K.P.; Ahlquist, D.A. Evidence against flat dysplasia as a regional field defect in small bowel adenocarcinoma associated with celiac sprue. Mayo Clin. Proc. 1997, 72, 320–322. [Google Scholar] [CrossRef]
- Schottenfeld, D.; Beebe-Dimmer, J.L.; Vigneau, F.D. The Epidemiology and Pathogenesis of Neoplasia in the Small Intestine. Ann. Epidemiol. 2009, 19, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Goodman, M.T.; Matsuno, R.K.; Shvetsov, Y.B. Racial and ethnic variation in the incidence of small-bowel cancer subtypes in the United States, 1995–2008. Dis. Colon Rectum 2013, 56, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Caio, G.; Volta, U.; Ursini, F.; Manfredini, R.; De Giorgio, R. Small bowel adenocarcinoma as a complication of celiac disease: Clinical and diagnostic features. BMC Gastroenterol. 2019, 19, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanoli, A.; Di Sabatino, A.; Martino, M.; Klersy, C.; Grillo, F.; Mescoli, C.; Nesi, G.; Volta, U.; Fornino, D.; Luinetti, O.; et al. Small bowel carcinomas in celiac or Crohn’s disease: Distinctive histophenotypic, molecular and histogenetic patterns. Mod. Pathol. 2017, 30, 1453–1466. [Google Scholar] [CrossRef]
- Grolleau, C.; Pote, N.M.; Guedj, N.S.; Zappa, M.; Theou-Anton, N.; Bouhnik, Y.; Panis, Y.; Cazals-Hatem, D.L. Small bowel adenocarcinoma complicating Crohn’s disease: A single-centre experience emphasizing the importance of screening for dysplasia. Virchows Arch. 2017, 471, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Hamilton, S.R. Genetic alterations in sporadic and Crohn’s-associated adenocarcinomas of the small intestine. Gastroenterology 1997, 113, 127–135. [Google Scholar] [CrossRef]
- Warth, A.; Kloor, M.; Schirmacher, P.; Bläker, H. Genetics and epigenetics of small bowel adenocarcinoma: The interactions of CIN, MSI, and CIMP. Mod. Pathol. 2011, 24, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Pozadzides, J.; Kopetz, S.; Wen, S.; Abbruzzese, J.L.; Wolff, R.A.; Wang, H. Immunophenotype and molecular characterisation of adenocarcinoma of the small intestine. Br. J. Cancer 2010, 102, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Bojesen, R.D.; Riis, L.B.; Høgdall, E.; Nielsen, O.H.; Jess, T. Inflammatory Bowel Disease and Small Bowel Cancer Risk, Clinical Characteristics, and Histopathology: A Population-Based Study. Clin. Gastroenterol. Hepatol. 2017, 15, 1900–1907.e2. [Google Scholar] [CrossRef]
- Rizzo, F.; Vanoli, A.; Sahnane, N.; Cerutti, R.; Trapani, D.; Rinaldi, A.; Sellitto, A.; Ciacci, C.; Volta, U.; Villanacci, V.; et al. Small-bowel carcinomas associated with celiac disease: Transcriptomic profiling shows predominance of microsatellite instability-immune and mesenchymal subtypes. Virchows Arch. 2020, 476, 711–723. [Google Scholar] [CrossRef]
- Schrock, A.B.; Devoe, C.E.; McWilliams, R.; Sun, J.; Aparicio, T.; Stephens, P.J.; Ross, J.S.; Wilson, R.; Miller, V.A.; Ali, S.M.; et al. Genomic profiling of small-bowel adenocarcinoma. JAMA Oncol. 2017, 3, 1546–1553. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, T.; Svrcek, M.; Zaanan, A.; Beohou, E.; Laforest, A.; Afchain, P.; Mitry, E.; Taieb, J.; Di Fiore, F.; Gornet, J.M.; et al. Small bowel adenocarcinoma phenotyping, a clinicobiological prognostic study. Br. J. Cancer 2013, 109, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Svrcek, M.; Piton, G.; Cosnes, J.; Beaugerie, L.; Vermeire, S.; Geboes, K.; Lemoine, A.; Cervera, P.; El-Murr, N.; Dumont, S.; et al. Small bowel adenocarcinomas complicating Crohn’s disease are associated with dysplasia: A pathological and molecular study. Inflamm. Bowel Dis. 2014, 20, 1584–1592. [Google Scholar] [CrossRef]
- Younes, N.; Fulton, N.; Tanaka, R.; Wayne, J.; Straus, F., 2nd; Kaplan, E. The presence of K-12 ras mutations in duodenal adenocarcinomas and the absence of ras mutations in other small bowel adenocarcinomas and carcinoid tumors. Cancer 1997, 79, 1804–1808. [Google Scholar] [CrossRef]
- Bläker, H.; Helmchen, B.; Bönisch, A.; Aulmann, S.; Penzel, R.; Otto, H.F.; Rieker, R.J. Mutational activation of the RAS-RAF-MAPK and the wnt pathway in small intestinal adenocarcinomas. Scand. J. Gastroenterol. 2004, 39, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, O.J.; Jang, K.T.; Bae, Y.K.; Chung, J.Y.; Eom, D.W.; Kim, J.M.; Yu, E.; Hong, S.M. Combined loss of e-cadherin and aberrant β-catenin protein expression correlates with a poor prognosis for small intestinal adenocarcinomas. Am. J. Clin. Pathol. 2013, 139, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Laforest, A.; Aparicio, T.; Zaanan, A.; Silva, F.P.; Didelot, A.; Desbeaux, A.; Le Corre, D.; Benhaim, L.; Pallier, K.; Aust, D.; et al. ERBB2 gene as a potential therapeutic target in small bowel adenocarcinoma. Eur. J. Cancer 2014, 50, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Perzin, K.H.; Bridge, M.F. Adenomas of the small intestine: A clinicopathologic review of 51 cases and a study of their relationship to carcinoma. Cancer 1981, 48, 799–819. [Google Scholar] [CrossRef]
- Genta, R.M.; Feagins, L.A. Advanced precancerous lesions in the small bowel mucosa. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Raghav, K.; Overman, M.J. Small bowel adenocarcinomas—existing evidence and evolving paradigms. Nat. Rev. Clin. Oncol. 2013, 10, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Tomba, C.; Sidhu, R.; Sanders, D.S.; Mooney, P.D.; Branchi, F.; Locatelli, M.; Roncoroni, L.; Conte, D.; Bardella, M.T.; Elli, L. Celiac Disease and Double-Balloon Enteroscopy: What Can We Achieve? The Experience of 2 European Tertiary Referral Centers. J. Clin. Gastroenterol. 2016, 50, 313–317. [Google Scholar] [CrossRef]
- Tomba, C.; Elli, L.; Bardella, M.T.; Soncini, M.; Contiero, P.; Roncoroni, L.; Locatelli, M.; Conte, D. Enteroscopy for the early detection of small bowel tumours in at-risk celiac patients. Dig. Liver Dis. 2014, 46, 400–404. [Google Scholar] [CrossRef]
- Overman, M.J.; Hu, C.-Y.; Kopetz, S.; Abbruzzese, J.L.; Wolff, R.A.; Chang, G.J. A Population-Based Comparison of Adenocarcinoma of the Large and Small Intestine: Insights Into a Rare Disease. Ann. Surg. Oncol. 2012, 19, 1439–1445. [Google Scholar] [CrossRef]
- Dabaja, B.S.; Suki, D.; Pro, B.; Bonnen, M.; Ajani, J. Adenocarcinoma of the small bowel. Cancer 2004, 101, 518–526. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; McWilliams, R.R.; Donohue, J.H.; Quevedo, J.F. A single-institution experience with 491 cases of small bowel adenocarcinoma. Am. J. Surg. 2010, 199, 797–803. [Google Scholar] [CrossRef]
- Aparicio, T.; Henriques, J.; Manfredi, S.; Tougeron, D.; Bouché, O.; Pezet, D.; Piessen, G.; Coriat, R.; Zaanan, A.; Legoux, J.-L.; et al. Small bowel adenocarcinoma: Results from a nationwide prospective ARCAD-NADEGE cohort study of 347 patients. Int. J. Cancer 2020, 147, 967–977. [Google Scholar] [CrossRef]
- Veyrières, M.; Baillet, P.; Hay, J.M.; Fingerhut, A.; Bouillot, J.L.; Julien, M. Factors influencing long-term survival in 100 cases of small intestine primary adenocarcinoma. Am. J. Surg. 1997, 173, 237–239. [Google Scholar] [CrossRef]
- Palascak-Juif, V.; Bouvier, A.M.; Cosnes, J.; Flourié, B.; Bouché, O.; Cadiot, G.; Lémann, M.; Bonaz, B.; Denet, C.; Marteau, P.; et al. Small bowel adenocarcinoma in patients with Crohn’s disease compared with small bowel adenocarcinoma de novo. Inflamm. Bowel Dis. 2005, 11, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.K.; Fletcher, J.G.; Fidler, J.L.; Barlow, J.M.; Pruthi, S.; Loftus, E.V.; Pardi, D.S.; Smyrk, T.C.; Becker, B.D.; Pasha, S.F.; et al. Clinical characteristics and imaging features of small bowel adenocarcinomas in Crohn’s disease. Abdom. Imaging 2015, 40, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, P.; Arpa, G.; Grillo, F.; Klersy, C.; Sampietro, G.; Ardizzone, S.; Fociani, P.; Fiocca, R.; Latella, G.; Sessa, F.; et al. PD-L1 in small bowel adenocarcinoma is associated with etiology and tumor-infiltrating lymphocytes, in addition to microsatellite instability. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2020, 33, 1398–1409. [Google Scholar] [CrossRef]
- Vanoli, A.; Grillo, F.; Guerini, C.; Neri, G.; Arpa, G.; Klersy, C.; Nesi, G.; Giuffrida, P.; Sampietro, G.; Ardizzone, S.; et al. Prognostic Role of Mismatch Repair Status, Histotype and High-Risk Pathologic Features in Stage II Small Bowel Adenocarcinomas. Ann. Surg. Oncol. 2021, 28, 1167–1177. [Google Scholar] [CrossRef]
- Locher, C.; Batumona, B.; Afchain, P.; Carrère, N.; Samalin, E.; Cellier, C.; Aparicio, T.; Becouarn, Y.; Bedenne, L.; Michel, P.; et al. Small bowel adenocarcinoma: French intergroup clinical practice guidelines for diagnosis, treatments and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO). Dig. Liver Dis. 2018, 50, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Santini, D.; Fratto, M.E.; Spoto, C.; Russo, A.; Galluzzo, S.; Zoccoli, A.; Vincenzi, B.; Tonini, G. Cetuximab in small bowel adenocarcinoma: A new friend. Br. J. Cancer 2010, 103, 1305. [Google Scholar] [CrossRef]
- Falcone, R.; Roberto, M.; Filetti, M.; Anselmi, E.; Marchetti, P. Anti epidermal growth factor receptor therapy in small bowel adenocarcinoma. Medicine 2018, 97, e9672. [Google Scholar] [CrossRef]
- Gulhati, P.; Raghav, K.; Shroff, R.; Varadhachary, G.; Javle, M.; Qiao, W.; Wang, H.; Morris, J.; Wolff, R.; Overman, M.J. Phase II Study of Panitumumab in RAS Wild-Type Metastatic Adenocarcinoma of Small Bowel or Ampulla of Vater. Oncologist 2018, 23, 277. [Google Scholar] [CrossRef] [Green Version]
- Hamad, A.; Singhi, A.D.; Bahary, N.; McGrath, K.; Amarin, R.; Zeh, H.J.; Zureikat, A.H. Neoadjuvant treatment with trastuzumab and folfox induces a complete pathologic response in a metastatic ERBB2 (HER2)-Amplified Duodenal Cancer. JNCCN J. Natl. Compr. Cancer Netw. 2017, 15, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Lebwohl, B.; Stavsky, E.; Neugut, A.I.; Green, P.H.R. Risk of colorectal adenomas in patients with coeliac disease. Aliment. Pharmacol. Ther. 2010, 32, 1037–1043. [Google Scholar] [CrossRef] [Green Version]
- Landgren, A.M.; Landgren, O.; Gridley, G.; Dores, G.M.; Linet, M.S.; Morton, L.M. Autoimmune disease and subsequent risk of developing alimentary tract cancers among 4.5 million US male veterans. Cancer 2011, 117, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Volta, U.; Vincentini, O.; Quintarelli, F.; Felli, C.; Silano, M. Low risk of colon cancer in patients with celiac disease. Scand. J. Gastroenterol. 2014, 49, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.F.; West, J.; Ekbom, A.; Stephansson, O. Reduced risk of breast, endometrial and ovarian cancer in women with celiac disease. Int. J. Cancer 2012, 131, E244–E250. [Google Scholar] [CrossRef]
- Persson, I. Estrogens in the causation of breast, endometrial and ovarian cancers - evidence and hypotheses from epidemiological findings. J. Steroid Biochem. Mol. Biol. 2000, 74, 357–364. [Google Scholar] [CrossRef]
- Olén, O.; Montgomery, S.M.; Marcus, C.; Ekbom, A.; Ludvigsson, J.F. Coeliac disease and body mass index: A study of two Swedish general population-based registers. Scand. J. Gastroenterol. 2009, 44, 1198–1206. [Google Scholar] [CrossRef]
- Michels, K.B.; Ekbom, A. Caloric restriction and incidence of breast cancer. JAMA 2004, 291, 1226–1230. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, M.; Melbye, M.; Wohlfahrt, J.; Sørensen, T.I.A. Growth patterns and the risk of breast cancer in women. N. Engl. J. Med. 2004, 351, 1619–1626. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Ansved, P.; Fälth-Magnusson, K.; Hammersjö, J.-A.; Johansson, C.; Edvardsson, S.; Ljungkrantz, M.; Stenhammar, L.; Ludvigsson, J. Symptoms and signs have changed in Swedish children with coeliac disease. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J.F.; Fall, K.; Montgomery, S. Risk of prostate cancer in a population-based cohort of men with coeliac disease. Br. J. Cancer 2012, 106, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.; McBride, R.; McConnell, R.; Neugut, A.I.; Bhagat, G.; Green, P.H.R. Increased risk of papillary thyroid cancer in celiac disease. Dig. Dis. Sci. 2006, 51, 1875–1877. [Google Scholar] [CrossRef]
- Volta, U.; Vincentini, O.; Silano, M. Papillary cancer of thyroid in celiac disease. J. Clin. Gastroenterol. 2011, 45, e44–e46. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Lebwohl, B.; Kämpe, O.; Murray, J.A.; Green, P.H.; Ekbom, A. Risk of thyroid cancer in a nationwide cohort of patients with biopsy-verified celiac disease. Thyroid 2013, 23, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Lebwohl, B.; Eriksson, H.; Hansson, J.; Green, P.H.R.; Ludvigsson, J.F. Risk of cutaneous malignant melanoma in patients with celiac disease: A population-based study. J. Am. Acad. Dermatol. 2014, 71, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Pereyra, L.; Gonzalez, R.; Mohaidle, A.; Fischer, C.; Mella, J.M.; Panigadi, G.N.; Manazzoni, D.; Matoso, M.D.; Lasa, J.S.; Novillo, A.; et al. Risk of colorectal neoplasia in patients with celiac disease: A multicenter study. J. Crohns. Colitis 2013, 7, e672–e677. [Google Scholar] [CrossRef] [Green Version]
- Silano, M.; Volta, U.; De Vincenzi, A.; Dessì, M.; Vincenzi, M. De Effect of a gluten-free diet on the risk of enteropathy-associated T-cell lymphoma in celiac disease. Dig. Dis. Sci. 2008, 53, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Olén, O.; Askling, J.; Ludvigsson, J.F.; Hildebrand, H.; Ekbom, A.; Smedby, K.E. Coeliac disease characteristics, compliance to a gluten free diet and risk of lymphoma by subtype. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2011, 43, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Kelly, C.P.; Leffler, D.A. Gastrointestinal cancer in celiac disease: “the first days are the hardest days, don’t you worry anymore? Clin. Gastroenterol. Hepatol. 2012, 10, 4–6. [Google Scholar] [CrossRef] [PubMed]
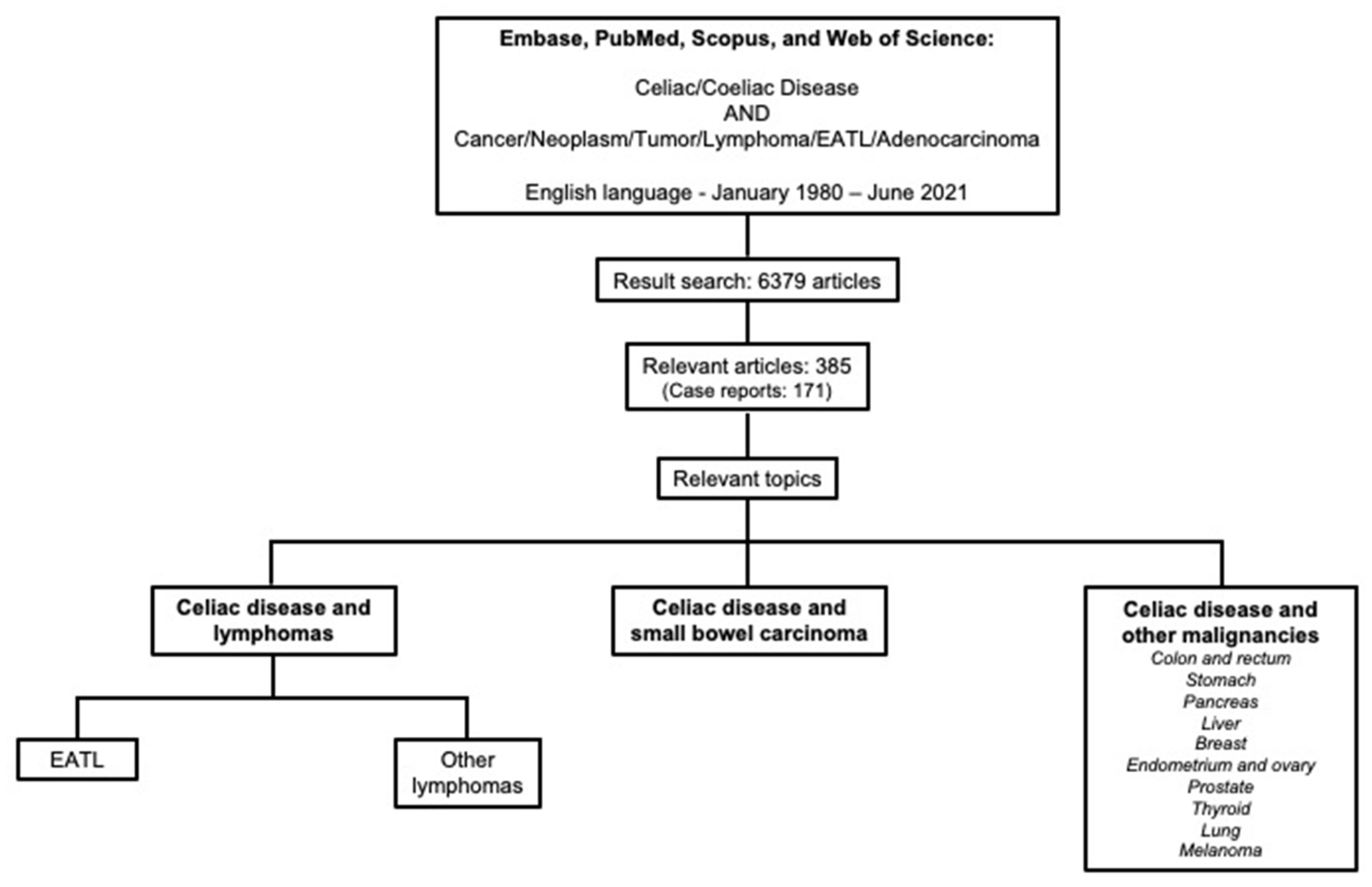
| Study | Year | Country | Study Design | CeD Cases—n | Lymphoma Cases—n | Main Findings |
|---|---|---|---|---|---|---|
| Holmes et al. [21] | 1989 | United Kingdom | Monocentric retrospective cohort study | 210 | 9 | Increased risk of NHL: SIR = 42.7 (95% CI 19.6–81.4) |
| Cottone et al. [50] | 1999 | Italy | Hospital-based retrospective cohort | 228 | 6 | Incidence of NHL was 3/100 against an expected incidence of 0.8 (p < 0.01) |
| Green et al. [51] | 2001 | USA | Nationwide cross-sectional survey | 1612 | 3 | Relative risk for the development of lymphoma of 300 (60–876) |
| Corrao et al. [8] | 2001 | Italy | Multicenter prospective cohort study | 1072 | 16 (death from NHL) | Increased risk of death from NHL: SMR = 69.3 (95% CI 40.7–112.6) |
| Askling et al. [23] | 2002 | Sweden | Population-based prospective cohort study | 11,019 | 44 | Increased risk of: NHL: SIR = 6.3 (95% CI 4.2–125) HL: SIR = 4.6 (95% CI 1.7–10) |
| Catassi et al. [43] | 2002 | Italy | Multicenter case–control study | 6 | 653 | CeD was associated with: NHL: OR = 3.1 (95% CI 1.3–7.6) Primary gut NHL: OR = 16.9 (95% CI 7.4–38.7) T-cell NHL: OR = 19.2 (95% CI 7.9–46.6) |
| Peters et al. [9] | 2003 | Sweden | Population-based retrospective cohort study | 10,032 | 22 | Excess of mortality for NHL: SMR = 11.4 (95% CI 7.8–16.0) |
| Green et al. [29] | 2003 | USA | Hospital-based prospective cohort study | 381 | 9 | Increased risk of NHL: standardized morbidity ratio = 9.1 (95% CI 4.7–13). |
| Howdle et al. [52] | 2003 | United Kingdom | Clinical registry-based cohort | 37 | 86 | 37/86 with lymphoma |
| Card et al. [24] | 2004 | United Kingdom | Population-based prospective cohort study | 869 | 12 | Peridiagnostic period: NHL: SIR = 20.94 (95% CI 6.8–48.86) Small bowel NHL: SIR = 358.8 (95% CI 74.01–1048.34) Postdiagnostic period: NHL: SIR = 5.8 (95% CI 1.58–14.86) Small bowel NHL: SIR = 40.51 (95% CI 1.03–225.68) |
| West et al. [25] | 2004 | United Kingdom | Population-based cohort study | 4732 | 23 | Increased risk of lymphoproliferative disease: aHR = 4.3 (95% CI 2.4–7.7) |
| Farré et al. [53] | 2004 | Spain | Multicenter case–control study | 5 | 298 | No risk of lymphoma detected in silent or recognized CeD patients: OR = 0.62 (95% CI 0.10–3.79) |
| Smedby et al. [44] | 2005 | Sweden | Population-based prospective cohort study | 11,650 | 56 | Increased risk of: NHL overall: SIR = 6.6 (95% CI 5.0–8.6) B-cell NHL: SIR = 2.2 (95% CI 1.3–3.6) T-cell NHL: SIR = 51 (95% CI 35–68) Intestinal NHL: SIR = 24 (16–35) Non-intestinal NHL: SIR = 3.6 (2.3–5.2) No increased risk of HL: SIR = 1.0 (95% CI 0.02–5.6) |
| Viljamaa et al. [11] | 2006 | Finland | Population-based prospective cohort study | 781 | 5 (4 EATL, 1 DLBCL) | Risk of NHL significantly increased: SIR = 3.2 (95% CI 1.0–7.5). Increased mortality risk from lymphoproliferative diseases: SMR = 4.12 (95% CI 1.66–8.51) |
| Smedby et al. [47] | 2006 | Denmark and Sweden | Population-based case–control study | 28 | 3055 | Increased risk of: NHL: OR = 2.1 (95% CI 1.0–4.8) Diffuse large B-cell NHL: OR = 2.8 (95% CI 1.0–8.0) T-cell NHL: OR = 17 (95% CI 6.3–46) |
| Mearin et al. [48] | 2006 | 10 European countries | Prospective, multicenter, case–control study | 66 | 1446 | Increased OR for NHL: 2.6 (95% CI 1.4–4.9) |
| Silano et al. [26] | 2007 | Italy | Population-based prospective cohort study | 1968 | 20 | Significant increase in NHL risk: SIR = 4.7 (95% CI 2.9–7.3) |
| Anderson et al. [30] | 2007 | United Kingdom | Population-based retrospective cohort study | 490 (EMA+) | 2 | No significant increased risk of NHL despite a raised SIR [7.47 (95% CI 0.00–17.83)] |
| Goldacre et al. [31] | 2008 | United Kingdom | Hospital-based retrospective cohort | 1997 | 11 | Increased risk of NHL: adjusted Rate Ratio = 3.28 (95% CI 1.49–6.28). No significant increase in HL: adjusted Rate Ratio = 5.07 (95% CI 0.61–18.7) |
| Lohi et al. [37] | 2009 | Finland | Population-based retrospective cohort study | 73 (EMA + subjects) | 2 | Increased mortality risk for lymphoma: RR = 9.51 (2.20–41.22) |
| Gao et al. [46] | 2009 | Sweden | Population-based case–control study | 54 in NHL patients 7 in HL patients 40 in controls | 37,869 NHL 8323 HL 236,408 controls | Increased risk of: NHL: OR = 5.35 (95% CI 3.56–8.06) HL: OR = 2.54 (95% CI 0.99– 6.56) |
| Anderson et al. [54] | 2009 | USA | Population-based case–control study | 25 | 33,721 | Borderline increased risk of NHL overall: OR = 1.5 (95% CI 0.9–2.5) Increased risk of: T-cell NHL: OR = 5.9 (95% CI 2.4–14) Marginal zone lymphoma: OR = 3.5 (95% CI 1.3–9.8) |
| Lohi et al. [32] | 2009 | Finland | Population-based retrospective cohort study | 73 (EMA + patients) | 2 | Increased risk of lymphoproliferative diseases: RR = 5.94 (95% CI 1.41–25.04) |
| Grainge et al. [10] | 2011 | United Kingdom | Population-based prospective cohort study | 1092 | 6 | Increased risk of death from NHL: SMR = 7.06 (95% CI 2.59–15.4) |
| Elfstrom et al. [45] | 2011 | Sweden | Population-based retrospective cohort study | 28,989 CD (Marsh 3) 13,140 inflammation (Marsh 1–2) 3711 positive serology | 289 | Increased risk of lymphoproliferative malignancy in: CeD patients: HR = 2.82 (95% CI 2.36–3.37) Inflammation: HR = 1.81 (95% CI 1.42–2.31) In patients with positive serology no increase in risk: HR = 0.97 (95% CI 0.44–2.14) |
| Grainge et al. [27] | 2012 | United Kingdom | Population-based retrospective cohort study | 435 | 14 | Increased risk of NHL: SIR = 12.0 (95% CI 6.55–20.1). The risk remained increased 15 years after CD diagnosis: SIR = 5.15 (95% CI 14.0–13.2) |
| Lebwohl et al. [55] | 2013 | Sweden | Population-based cohort study | 7625 | 53 | Increased risk of NHL: SIR = 2.81 (95% CI 2.10–3.67). In patients with persistent villous atrophy: SIR = 3.78 (95% CI 2.71–5.12) In patients with mucosal healing: SIR = 1.50 (95% CI 0.77–2.62) |
| Ilus et al. [56] | 2014 | Finland | Population-based prospective cohort study | 32,439 | 132 | Increased risk of NHL: SIR = 1.94 (95% CI 1.62–2.29) No increased risk of HL: SIR = 0.53 (95% CI 0.11–1.55) The SIR for NHL was increased (2.56) within 2 years from CeD diagnosis, but not at longer follow-up. |
| Abdul Sultan et al. [39] | 2015 | United Kingdom | Population-based retrospective cohort study | 10,825 | 26 | Mortality rate per 10,000 person years: 4.3 (2.9–6.3) in CeD vs. 1.4 (1.1–1.7) in controls. Patients with CeD had a 0.15% excess risk of dying from NHL up to 10 years post diagnosis. |
| van Gils et al. [49] | 2018 | Netherlands | Population-based, case–control study | 261 in lymphomas and GI carcinomas 282 in controls (melanoma and basal cell carcinoma) | 301,337 (lymphomas and GI carcinomas) 576,971 controls | Increased risk of T-cell lymphoma: RR = 35.8 (95% CI 27.1–47.4) |
| Quarpong et al. [34] | 2019 | United Kingdom | Population-based retrospective cohort study | 602 | 16 | SMR for lymphatic and hematopoietic malignancies: Overall = 5.16 (95% CI 2.95–8.38) Diagnosis at <15 years = 8.03 (95% CI 1.66–23) Diagnosis at ≥15 years = 4.77 (95% CI 2.54–8.16) |
| Koskinen et al. [40] | 2020 | Finland | Population-based cohort study | 12,803 | 44 | Increased risk of dying from lymphoproliferative diseases: HR = 2.36 (95% CI 1.65–3.39). HR decreased but remained significant after exclusion of the first 2 years of follow-up. |
| Lebwohl et al. [28] | 2021 | Sweden | Population-based cohort study | 47,241 | 445 hematologic cancers 392 lymphoproliferative cancers | Increased risk of: Hematologic cancers: HR = 1.90 (95% CI 1.70–2.13). Lymphoproliferative cancers: HR = 2.20 (95% CI 1.94–2.49). Greater risk persists also excluding the first-year follow-up. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelizzaro, F.; Marsilio, I.; Fassan, M.; Piazza, F.; Barberio, B.; D’Odorico, A.; Savarino, E.V.; Farinati, F.; Zingone, F. The Risk of Malignancies in Celiac Disease—A Literature Review. Cancers 2021, 13, 5288. https://doi.org/10.3390/cancers13215288
Pelizzaro F, Marsilio I, Fassan M, Piazza F, Barberio B, D’Odorico A, Savarino EV, Farinati F, Zingone F. The Risk of Malignancies in Celiac Disease—A Literature Review. Cancers. 2021; 13(21):5288. https://doi.org/10.3390/cancers13215288
Chicago/Turabian StylePelizzaro, Filippo, Ilaria Marsilio, Matteo Fassan, Francesco Piazza, Brigida Barberio, Anna D’Odorico, Edoardo V. Savarino, Fabio Farinati, and Fabiana Zingone. 2021. "The Risk of Malignancies in Celiac Disease—A Literature Review" Cancers 13, no. 21: 5288. https://doi.org/10.3390/cancers13215288
APA StylePelizzaro, F., Marsilio, I., Fassan, M., Piazza, F., Barberio, B., D’Odorico, A., Savarino, E. V., Farinati, F., & Zingone, F. (2021). The Risk of Malignancies in Celiac Disease—A Literature Review. Cancers, 13(21), 5288. https://doi.org/10.3390/cancers13215288