
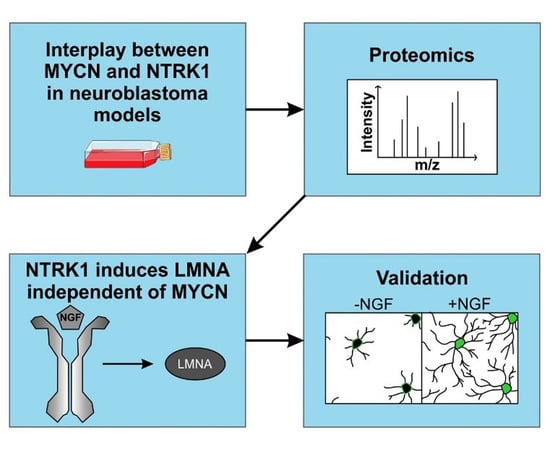
NTRK1/TrkA Signaling in Neuroblastoma Cells Induces Nuclear Reorganization and Intra-Nuclear Aggregation of Lamin A/C

 ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Validation of NTRK1/TrkA Expression and Activation in Four Established Human Neuroblastoma Cell Lines with Different MYCN Backgrounds
2.2. Proteome and Phosphoproteome Analyses Provide Time-Resolved Dynamics of NGF-NTRK1 Signaling in IMR5 Cells
2.3. NTRK1/TrkA Activation Modulates Cellular Pathways Related to Nuclear Lamina Integrity
2.4. NTRK1/TrkA Signaling Leads to LMNA Translocation and Aggregation in Nuclear Foci
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Lines
4.2. Western Blot Analyses
4.3. Activation of Ectopically Expressed NTRK1/TrkA and Preparation of Lysates for Proteome Analyses
4.4. Phosphopeptide Enrichment
4.5. LC-MS/MS Analysis
4.6. Mass Spectrometry Data Analysis
4.7. Statistical Analysis
4.8. Pathway Analysis
4.9. Immunofluorescence Staining and Quantitative Image Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Hanemaaijer, E.S.; Margaritis, T.; Sanders, K.; Bos, F.L.; Candelli, T.; Al-Saati, H.; van Noesel, M.M.; Meyer-Wentrup, F.A.G.; van de Wetering, M.; Holstege, F.C.P.; et al. Single-cell atlas of developing murine adrenal gland reveals relation of Schwann cell precursor signature to neuroblastoma phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2022350118. [Google Scholar] [CrossRef]
- Kildisiute, G.; Kholosy, W.M.; Young, M.D.; Roberts, K.; Elmentaite, R.; van Hooff, S.R.; Pacyna, C.N.; Khabirova, E.; Piapi, A.; Thevanesan, C.; et al. Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci. Adv. 2021, 7, eabd3311. [Google Scholar] [CrossRef]
- Schulte, J.H.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. Mycn and ALK-F1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef]
- Althoff, K.; Beckers, A.; Bell, E.; Nortmeyer, M.; Thor, T.; Sprussel, A.; Lindner, S.; De Preter, K.; Florin, A.; Heukamp, L.C.; et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015, 34, 3357–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazes, A.; Lopez-Delisle, L.; Tsarovina, K.; Pierre-Eugene, C.; De Preter, K.; Peuchmaur, M.; Nicolas, A.; Provost, C.; Louis-Brennetot, C.; Daveau, R.; et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 2688–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, H.; van Sluis, P.; de Kraker, J.; Bokkerink, J.; Egeler, M.; Laureys, G.; Slater, R.; Westerveld, A.; Voute, P.A.; Versteeg, R. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 1996, 334, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Theissen, J.; Oberthuer, A.; Hombach, A.; Volland, R.; Hertwig, F.; Fischer, M.; Spitz, R.; Zapatka, M.; Brors, B.; Ortmann, M.; et al. Chromosome 17/17q gain and unaltered profiles in high resolution array-CGH are prognostically informative in neuroblastoma. Genes Chromosomes Cancer 2014, 53, 639–649. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Pajtler, K.W.; Mahlow, E.; Odersky, A.; Lindner, S.; Stephan, H.; Bendix, I.; Eggert, A.; Schramm, A.; Schulte, J.H. Neuroblastoma in dialog with its stroma: NTRK1 is a regulator of cellular cross-talk with Schwann cells. Oncotarget 2014, 5, 11180–11192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Rebmann, V.; Lindemann, M.; Schulte, J.H.; Schulte, S.; Stauder, M.; Leuschner, I.; Schmid, K.W.; Kohl, U.; Schramm, A.; et al. Expression of NTRK1/TrkA affects immunogenicity of neuroblastoma cells. Int. J. Cancer 2013, 133, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Iraci, N.; Diolaiti, D.; Papa, A.; Porro, A.; Valli, E.; Gherardi, S.; Herold, S.; Eilers, M.; Bernardoni, R.; Della Valle, G.; et al. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TrkA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res. 2011, 71, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Schowe, B.; Fielitz, K.; Heilmann, M.; Martin, M.; Marschall, T.; Koster, J.; Vandesompele, J.; Vermeulen, J.; de Preter, K.; et al. Exon-level expression analyses identify MYCN and NTRK1 as major determinants of alternative exon usage and robustly predict primary neuroblastoma outcome. Br. J. Cancer 2012, 107, 1409–1417. [Google Scholar] [CrossRef]
- Emdal, K.B.; Pedersen, A.K.; Bekker-Jensen, D.B.; Tsafou, K.P.; Horn, H.; Lindner, S.; Schulte, J.H.; Eggert, A.; Jensen, L.J.; Francavilla, C.; et al. Temporal proteomics of NGF-TrkA signaling identifies an inhibitory role for the E3 ligase Cbl-b in neuroblastoma cell differentiation. Sci. Signal. 2015, 8, ra40. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Schramm, A.; Klein-Hitpass, L.; Klenk, M.; Wessels, H.; Hauffa, B.P.; Eils, J.; Eils, R.; Brodeur, G.M.; Schweigerer, L.; et al. Microarray analysis reveals differential gene expression patterns and regulation of single target genes contributing to the opposing phenotype of TrkA- and TrkB-expressing neuroblastomas. Oncogene 2005, 24, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Graaf, E.L.; Giansanti, P.; Altelaar, A.F.; Heck, A.J. Single-step enrichment by Ti4+-IMAC and label-free quantitation enables in-depth monitoring of phosphorylation dynamics with high reproducibility and temporal resolution. Mol. Cell. Proteom. MCP 2014, 13, 2426–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumyatsky, G.P.; Malleret, G.; Shin, R.M.; Takizawa, S.; Tully, K.; Tsvetkov, E.; Zakharenko, S.S.; Joseph, J.; Vronskaya, S.; Yin, D.; et al. Stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell 2005, 123, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, S.; Maples, P.B.; Senzer, N.; Nemunaitis, J. Stathmin 1: A novel therapeutic target for anticancer activity. Expert Rev. Anticancer Ther. 2008, 8, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Machado-Neto, J.A.; Saad, S.T.; Traina, F. Stathmin 1 in normal and malignant hematopoiesis. BMB Rep. 2014, 47, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takekoshi, K.; Nomura, F.; Isobe, K.; Motooka, M.; Nammoku, T.; Nakai, T. Identification and initial characterization of stathmin by the differential display method in nerve growth factor-treated PC12 cells. Eur. J. Endocrinol. 1998, 138, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Ohkawa, N.; Fujitani, K.; Tokunaga, E.; Furuya, S.; Inokuchi, K. The microtubule destabilizer stathmin mediates the development of dendritic arbors in neuronal cells. J. Cell Sci. 2007, 120, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Po’uha, S.T.; Le Grand, M.; Brandl, M.B.; Gifford, A.J.; Goodall, G.J.; Khew-Goodall, Y.; Kavallaris, M. Stathmin levels alter PTPN14 expression and impact neuroblastoma cell migration. Br. J. Cancer 2020, 122, 434–444. [Google Scholar] [CrossRef]
- Fife, C.M.; Sagnella, S.M.; Teo, W.S.; Po’uha, S.T.; Byrne, F.L.; Yeap, Y.Y.; Ng, D.C.H.; Davis, T.P.; McCarroll, T.A.; Kavallaris, M. Stathmin mediates neuroblastoma metastasis in a tubulin-independent manner via RhoA/ROCK signaling and enhanced transendothelial migration. Oncogene 2017, 36, 501–511. [Google Scholar] [CrossRef]
- Guglielmi, L.; Nardella, M.; Musa, C.; Iannetti, I.; Arisi, I.; D’Onofrio, M.; Storti, A.; Valentini, A.; Cacci, E.; Biagioni, S.; et al. Lamin A/C is Required for ChAT-Dependent Neuroblastoma Differentiation. Mol. Neurobiol. 2017, 54, 3729–3744. [Google Scholar] [CrossRef]
- Nardella, M.; Guglielmi, L.; Musa, C.; Iannetti, I.; Maresca, G.; Amendola, D.; Porru, M.; Carico, E.; Sessa, G.; Camerlingo, R.; et al. Down-regulation of the Lamin A/C in neuroblastoma triggers the expansion of tumor initiating cells. Oncotarget 2015, 6, 32821–32840. [Google Scholar] [CrossRef] [PubMed]
- Rauschert, I.; Aldunate, F.; Preussner, J.; Arocena-Sutz, M.; Peraza, V.; Looso, M.; Benech, J.C.; Agrelo, R. Promoter hypermethylation as a mechanism for Lamin A/C silencing in a subset of neuroblastoma cells. PLoS ONE 2017, 12, e0175953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moir, R.D.; Spann, T.P.; Herrmann, H.; Goldman, R.D. Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J. Cell Biol. 2000, 149, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Lessard, F.; Acevedo-Aquino, M.; Vernier, M.; Tsantrizos, Y.S.; Ferbeyre, G. Mutant lamin A links prophase to a p53 independent senescence program. Cell Cycle 2015, 14, 2408–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwermer, M.; Lee, S.; Köster, J.; van Maerken, T.; Stephan, H.; Eggert, A.; Morik, K.; Schulte, J.H.; Schramm, A. Sensitivity to cdk1-inhibition is modulated by p53 status in preclinical models of embryonal tumors. Oncotarget 2015, 6, 15425–15435. [Google Scholar] [CrossRef]
- Tjaden, B.; Baum, K.; Marquardt, V.; Simon, M.; Trajkovic-Arsic, M.; Kouril, T.; Siebers, B.; Lisec, J.; Siveke, J.T.; Schulte, J.H.; et al. N-Myc-induced metabolic rewiring creates novel therapeutic vulnerabilities in neuroblastoma. Sci. Rep. 2020, 10, 7157. [Google Scholar] [CrossRef]
- Megger, D.A.; Bracht, T.; Kohl, M.; Ahrens, M.; Naboulsi, W.; Weber, F.; Hoffmann, A.C.; Stephan, C.; Kuhlmann, K.; Eisenacher, M.; et al. Proteomic differences between hepatocellular carcinoma and nontumorous liver tissue investigated by a combined gel-based and label-free quantitative proteomics study. Mol. Cell. Proteom. MCP 2013, 12, 2006–2020. [Google Scholar] [CrossRef] [Green Version]
- Stepath, M.; Zulch, B.; Maghnouj, A.; Schork, K.; Turewicz, M.; Eisenacher, M.; Hahn, S.; Sitek, B.; Bracht, T. Systematic Comparison of Label-Free, SILAC, and TMT Techniques to Study Early Adaption toward Inhibition of EGFR Signaling in the Colorectal Cancer Cell Line DiFi. J. Proteome Res. 2020, 19, 926–937. [Google Scholar] [CrossRef]
- Stepath, M.; Bracht, T. Application of SILAC Labeling in Phosphoproteomics Analysis. Methods Mol. Biol. 2021, 2228, 167–183. [Google Scholar]
- Hogrebe, A.; von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.T.; Kelstrup, C.D.; Olsen, J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Al-Refae, K.; Krysztofiak, A.; Jendrossek, V. The Focinator v2-0—Graphical interface, four channels, colocalization analysis and cell phase identification. Radiat. Res. 2017, 188, 114–120. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funke, L.; Bracht, T.; Oeck, S.; Schork, K.; Stepath, M.; Dreesmann, S.; Eisenacher, M.; Sitek, B.; Schramm, A. NTRK1/TrkA Signaling in Neuroblastoma Cells Induces Nuclear Reorganization and Intra-Nuclear Aggregation of Lamin A/C. Cancers 2021, 13, 5293. https://doi.org/10.3390/cancers13215293
Funke L, Bracht T, Oeck S, Schork K, Stepath M, Dreesmann S, Eisenacher M, Sitek B, Schramm A. NTRK1/TrkA Signaling in Neuroblastoma Cells Induces Nuclear Reorganization and Intra-Nuclear Aggregation of Lamin A/C. Cancers. 2021; 13(21):5293. https://doi.org/10.3390/cancers13215293
Chicago/Turabian StyleFunke, Lukas, Thilo Bracht, Sebastian Oeck, Karin Schork, Markus Stepath, Sabine Dreesmann, Martin Eisenacher, Barbara Sitek, and Alexander Schramm. 2021. "NTRK1/TrkA Signaling in Neuroblastoma Cells Induces Nuclear Reorganization and Intra-Nuclear Aggregation of Lamin A/C" Cancers 13, no. 21: 5293. https://doi.org/10.3390/cancers13215293
APA StyleFunke, L., Bracht, T., Oeck, S., Schork, K., Stepath, M., Dreesmann, S., Eisenacher, M., Sitek, B., & Schramm, A. (2021). NTRK1/TrkA Signaling in Neuroblastoma Cells Induces Nuclear Reorganization and Intra-Nuclear Aggregation of Lamin A/C. Cancers, 13(21), 5293. https://doi.org/10.3390/cancers13215293