
Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance?


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
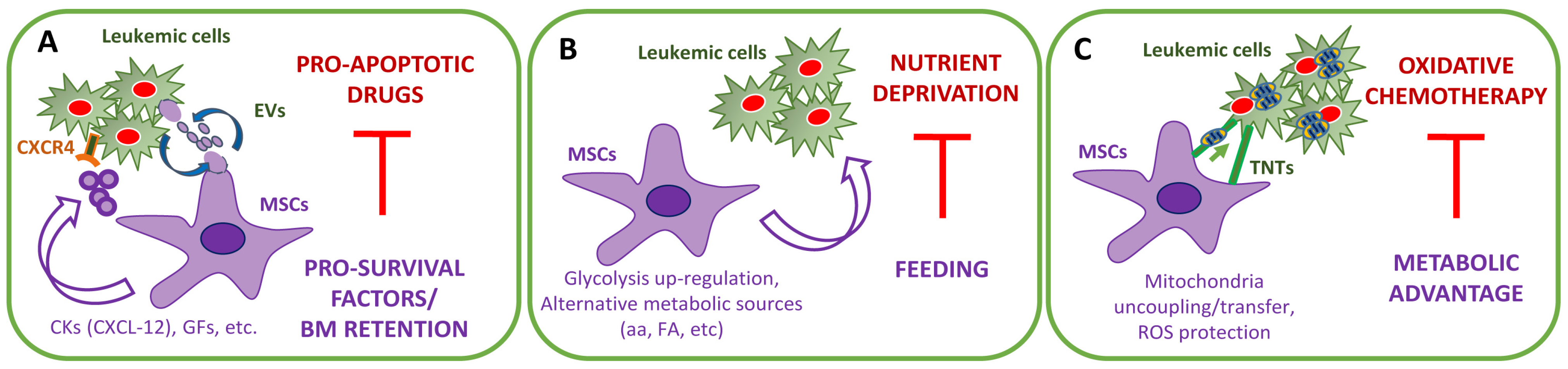
2. The Stromal-Dependent Anti-Apoptotic Effects
2.1. MSC-Dependent Pro-Survival Pathways
2.2. The Emerging Role of Extracellular Vesicles
3. The Stromal-Dependent Metabolic Reprogramming
3.1. Glucose Addiction
3.2. Biofuels Alternative to Glucose: Glutamine, Asparagine, and Cysteine
3.3. Biofuels Alternative to Glucose: Fatty Acids
- Leukemia cells prompt their microenvironment to increase FA production and availability.
- FAO promotes AML cell survival.
- BM-adipocytes impair the efficacy of anti-leukemia drugs.
3.4. Glycolysis versus OXPHOS
3.5. ROS and Redox Metabolism
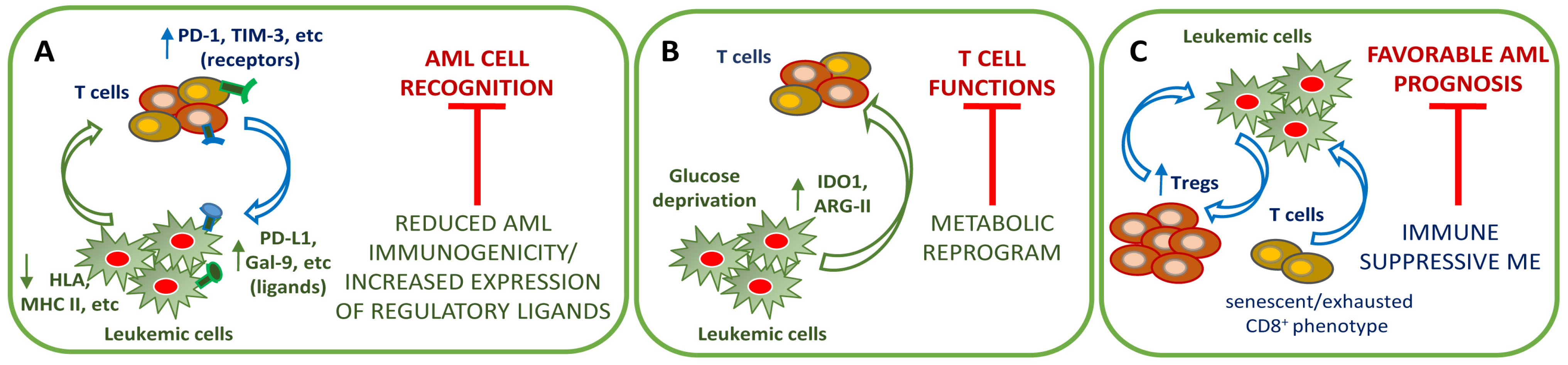
4. The Immune BM Microenvironment in Chemoresistance
4.1. AML-Mediated Mechanism of Immune Evasion
4.2. Immune System Dysregulation
4.3. Immune System Modulation as a Chance for Therapy
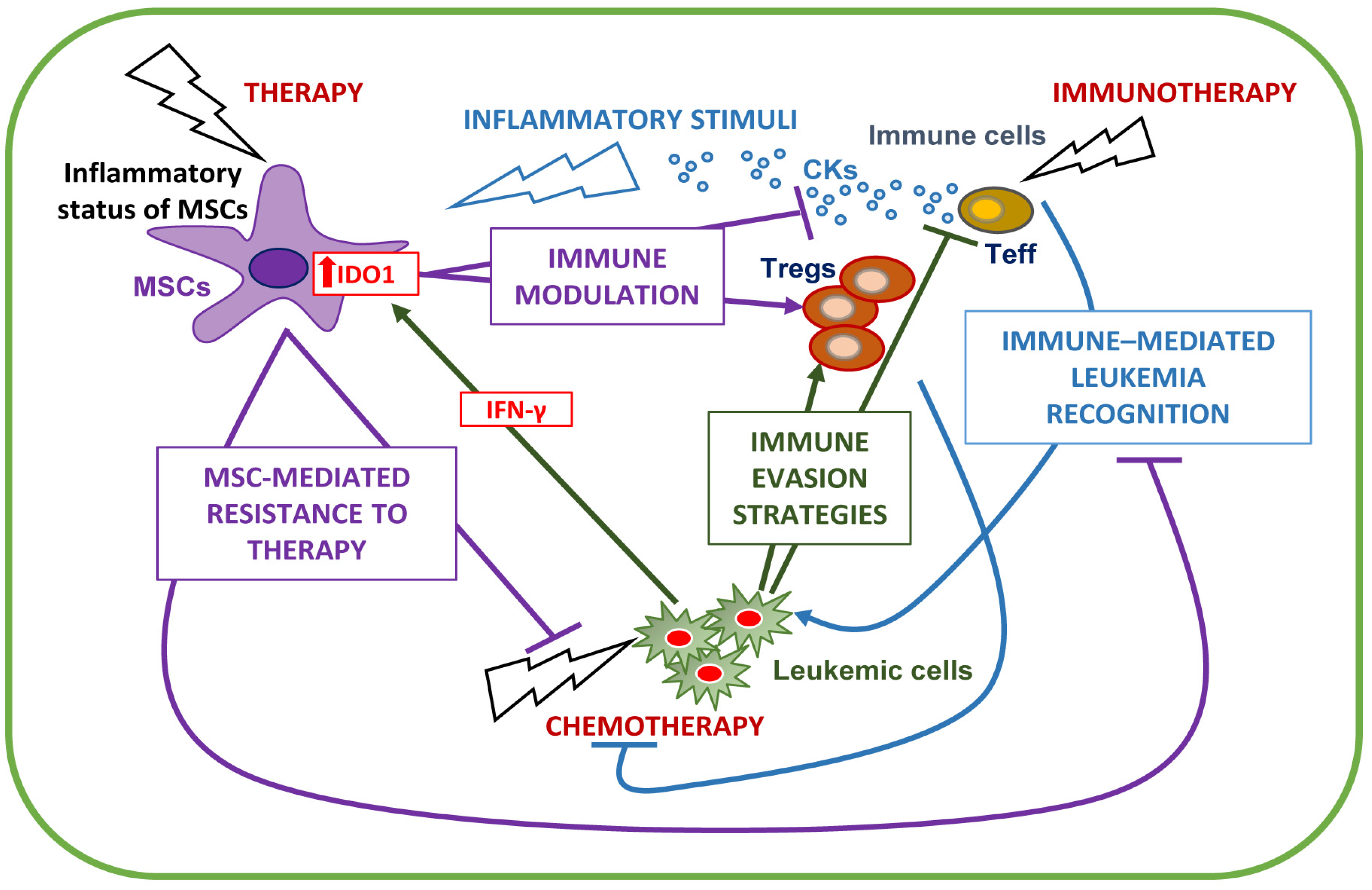
5. Stromal/Immune BM Microenvironment Interplay
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamashita, M.; Dellorusso, P.V.; Olson, O.C.; Passegué, E. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat. Rev. Cancer 2020, 20, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Récher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J. Clin. Investig. 2011, 121, 384–395. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Bendall, L.J.; Daniel, A.; Kortlepel, K.; Gottlieb, D.J. Bone marrow adherent layers inhibit apoptosis of acute myeloid leukemia cells. Exp. Hematol. 1994, 22, 1252–1260. [Google Scholar]
- Garrido, S.M.; Appelbaum, F.R.; Willman, C.L.; Banker, D.E. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp. Hematol. 2001, 29, 448–457. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norgaard, J.M.; Langkjer, S.T.; Palshof, T.; Clausen, N.; Pedersen, B.; Hokland, P. Relation of blast cell survival and proliferation to chemotherapy resistance in AML. Br. J. Haematol. 1996, 93, 888–897. [Google Scholar] [CrossRef]
- Banker, D.E.; Groudine, M.; Norwood, T.; Appelbaum, F.R. Measurement of spontaneous and therapeutic agent-induced apoptosis with BCL-2 protein expression in acute myeloid leukemia. Blood 1997, 89, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Maung, Z.T.; Hogarth, L.; Reid, M.M.; Proctor, S.J.; Hamilton, P.J.; Hall, A.G. Raised intracellular glutathione levels correlate with in vitro resistance to cytotoxic drugs in leukaemic cells from patients with acute lymphoblastic leukemia. Leukemia 1994, 8, 1487–1491. [Google Scholar]
- Golay, J.; Cuppini, L.; Leoni, F.; Mico, C.; Barbui, V.; Domenghini, M.; Lombardi, L.; Neri, A.; Barbui, A.M.; Salvi, A.; et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia 2007, 21, 1892–1900. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.Y.; Afanasiev, B.V.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabe, Y.; Jin, L.; Tsutsumi-Ishii, Y.; Xu, Y.; McQueen, T.; Priebe, W.; Mills, G.B.; Ohsaka, A.; Nagaoka, I.; Andreeff, M.; et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res. 2007, 67, 684–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreeff, M.; Jiang, S.; Zhang, X.; Konopleva, M.; Estrov, Z.; Snell, V.E.; Xie, Z.; Okcu, M.F.; Sanchez-Williams, G.; Dong, J.; et al. Expression of Bcl-2-related genes in normal and AML progenitors: Changes induced by chemotherapy and retinoic acid. Leukemia 1999, 13, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Karjalainen, R.; Pemovska, T.; Popa, M.; Liu, M.; Javarappa, K.K.; Majumder, M.M.; Yadav, B.; Tamborero, D.; Tang, J.; Bychkov, D.; et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood 2017, 130, 789–802. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, R.; Ruvolo, V.R.; Wei, J.; Konopleva, M.; Reed, J.C.; Pellecchia, M.; Andreeff, M.; Ruvolo, P.P. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (−)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood 2015, 126, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [CrossRef] [Green Version]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; San Martin, M.; Pop-Damkov, P.; Su, N.; Roamio Franklin, V.N.; Sekhar Reddy Chilamakuri, C.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef] [PubMed]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moujalled, D.M.; Pomilio, G.; Ghiurau, C.; Ivey, A.; Salmon, J.; Rijal, S.; Macraild, S.; Zhang, L.; Teh, T.C.; Tiong, I.S.; et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 2019, 33, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Warmoes, M.; Lorenzi, P.L.; Mak, D.; Ruvolo, V.; Tan, L.; Cidado, J.; Drew, L.; et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Cancilla, D.; Rettig, M.P.; DiPersio, J.F. Targeting CXCR4 in AML and ALL. Front. Oncol. 2020, 10, 1672. [Google Scholar] [CrossRef]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Möhle, R.; Bautz, F.; Rafii, S.; Moore, M.A.; Brugger, W.; Kanz, L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 1998, 91, 4523–4530. [Google Scholar] [CrossRef]
- Zeng, Z.; Samudio, I.J.; Munsell, M.; An, J.; Huang, Z.; Estey, E.; Andreeff, M.; Konopleva, M. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol. Cancer Ther. 2006, 5, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.L.; Rinella, S.P.; Hess, N.J.; Turicek, D.P.; Kabakov, S.A.; Zhu, F.; Bouchlaka, M.N.; Olson, S.L.; Cho, M.M.; Quamine, A.E.; et al. CXCR4 allows T cell acute lymphoblastic leukemia to escape from JAK1/2 and BCL2 inhibition through CNS infiltration. Leuk. Lymphoma 2021, 62, 1167–1177. [Google Scholar] [CrossRef]
- Caivano, A.; La Rocca, F.; Laurenzana, I.; Trino, S.; De Luca, L.; Lamorte, D.; Del Vecchio, L.; Musto, P. Extracellular Vesicles in Hematological Malignancies: From Biology to Therapy. Int. J. Mol. Sci. 2017, 18, 1183. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.S.; Jeong, E.; Boyiadzis, M.; Whiteside, T.L. Increased small extracellular vesicle secretion after chemotherapy via upregulation of cholesterol metabolism in acute myeloid leukaemia. J. Extracell. Vesicles 2020, 9, 1800979. [Google Scholar] [CrossRef]
- Trino, S.; Lamorte, D.; Caivano, A.; De Luca, L.; Sgambato, A.; Laurenzana, I. Clinical relevance of extracellular vesicles in hematological neoplasms: From liquid biopsy to cell biopsy. Leukemia 2021, 35, 661–678. [Google Scholar] [CrossRef]
- Nehrbas, J.; Butler, J.T.; Chen, D.W.; Kurre, P. Extracellular Vesicles and Chemotherapy Resistance in the AML Microenvironment. Front. Oncol. 2020, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef]
- Lyu, T.; Wang, Y.; Li, D.; Yang, H.; Qin, B.; Zhang, W.; Li, Z.; Cheng, C.; Zhang, B.; Guo, R.; et al. Exosomes from BM-MSCs promote acute myeloid leukemia cell proliferation, invasion and chemoresistance via upregulation of S100A4. Exp. Hematol. Oncol. 2021, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Viola, S.; Traer, E.; Huan, J.; Hornick, N.I.; Tyner, J.W.; Agarwal, A.; Loriaux, M.; Johnstone, B.; Kurre, P. Alterations in acute myeloid leukaemia bone marrow stromal cell exosome content coincide with gains in tyrosine kinase inhibitor resistance. Br. J. Haematol. 2016, 172, 983–986. [Google Scholar] [CrossRef]
- Ji, D.; He, Y.; Lu, W.; Rong, Y.; Li, F.; Huang, X.; Huang, R.; Jiang, Y.; Chen, G. Small-sized extracellular vesicles (EVs) derived from acute myeloid leukemia bone marrow mesenchymal stem cells transfer miR-26a-5p to promote acute myeloid leukemia cell proliferation, migration, and invasion. Hum. Cell 2021, 34, 965–976. [Google Scholar] [CrossRef]
- Xu, Y.C.; Lin, Y.S.; Zhang, L.; Lu, Y.; Sun, Y.L.; Fang, Z.G.; Li, Z.Y.; Fan, R.F. MicroRNAs of bone marrow mesenchymal stem cell-derived exosomes regulate acute myeloid leukemia cell proliferation and apoptosis. Chin. Med. J. (Engl.) 2020, 133, 2829–2839. [Google Scholar] [CrossRef]
- Zhang, F.; Lu, Y.; Wang, M.; Zhu, J.; Li, J.; Zhang, P.; Yuan, Y.; Zhu, F. Exosomes derived from human bone marrow mesenchymal stem cells transfer miR-222-3p to suppress acute myeloid leukemia cell proliferation by targeting IRF2/INPP4B. Mol. Cell. Probes 2020, 51, 101513. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia- permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Doron, B.; Abdelhamed, S.; Butler, J.T.; Hashmi, S.K.; Horton, T.M.; Kurre, P. Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 2019, 33, 918–930. [Google Scholar] [CrossRef]
- Gargiulo, E.; Morande, P.E.; Largeot, A.; Moussay, E.; Paggetti, J. Diagnostic and Therapeutic Potential of Extracellular Vesicles in B-Cell Malignancies. Front. Oncol. 2020, 10, 580874. [Google Scholar] [CrossRef] [PubMed]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Koch, R.; Aung, T.; Vogel, D.; Chapuy, B.; Wenzel, D.; Becker, S.; Sinzig, U.; Venkataramani, V.; von Mach, T.; Jacob, R.; et al. Nuclear Trapping through Inhibition of Exosomal Export by Indomethacin Increases Cytostatic Efficacy of Doxorubicin and Pixantrone. Clin. Cancer Res. 2016, 22, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Tabe, Y.; Lorenzi, P.L.; Konopleva, M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood 2019, 134, 1014–1023. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M.; Andreeff, M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front. Oncol. 2020, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.; Xuan, L.I.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Akers, L.J.; Fang, W.; Levy, A.G.; Franklin, A.R.; Huang, P.; Zweidler-McKay, P.A. Targeting glycolysis in leukemia: A novel inhibitor 3-BrOP in combination with rapamycin. Leuk. Res. 2011, 35, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Vander Heiden, M.G.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Fiegl, M.; McQueen, T.; Clise-Dwyer, K.; Andreeff, M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008, 68, 5198–5205. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Qorraj, M.; Büttner, M.; Klein, F.A.; Saul, D.; Aigner, M.; Huber, W.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia 2016, 30, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Long, Z.J.; Wang, L.X.; Zheng, F.M.; Fang, Z.G.; Yan, M.; Xu, D.F.; Chen, J.J.; Wang, S.W.; Lin, D.J.; et al. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Mol. Cancer Res. 2013, 11, 1326–1336. [Google Scholar] [CrossRef] [Green Version]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, Y.X.; Tsao, T.; Qiu, Y.; Kornblau, S.M.; Baggerly, K.A.; Liu, W.; Jessen, K.; Liu, Y.; Kantarjian, H.; et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012, 120, 2679–2689. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuera, G.A.; Schop, D.; Spitters, T.W.; van Dijkhuizen-Radersma, R.; Bracke, M.; de Bruijn, J.D.; Martens, D.; Karperien, M.; van Boxtel, A.; van Blitterswijk, C.A. Patterns of amino acid metabolism by proliferating human mesenchymal stem cells. Tissue Eng. Part A 2012, 18, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef] [Green Version]
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.e396. [Google Scholar] [CrossRef]
- Parmentier, J.H.; Maggi, M.; Tarasco, E.; Scotti, C.; Avramis, V.I.; Mittelman, S.D. Glutaminase activity determines cytotoxicity of L-asparaginases on most leukemia cell lines. Leuk. Res. 2015, 39, 757–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2019, 35, 333–335. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019, 134, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutter, J.; Huang, Y.; Marovca, B.; Vonderheit, A.; Grotzer, M.A.; Eckert, C.; Cario, G.; Wollscheid, B.; Horvath, P.; Bornhauser, B.C.; et al. Image-based RNA interference screening reveals an individual dependence of acute lymphoblastic leukemia on stromal cysteine support. Oncotarget 2014, 5, 11501–11512. [Google Scholar] [CrossRef] [Green Version]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corradi, G.; Baldazzi, C.; Očadlíková, D.; Marconi, G.; Parisi, S.; Testoni, N.; Finelli, C.; Cavo, M.; Curti, A.; Ciciarello, M. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res. Ther. 2018, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Weickert, M.-T.; Hecker, J.S.; Buck, M.C.; Schreck, C.; Rivière, J.; Schiemann, M.; Schallmoser, K.; Bassermann, F.; Strunk, D.; Oostendorp, R.A.J.; et al. Bone marrow stromal cells from MDS and AML patients show increased adipogenic potential with reduced Delta-like-1 expression. Sci. Rep. 2021, 11, 5944. [Google Scholar] [CrossRef]
- Justesen, J.; Stenderup, K.; Ebbesen, E.N.; Mosekilde, L.; Steiniche, T.; Kassem, M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001, 2, 165–171. [Google Scholar] [CrossRef]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2020, 34, 391–403. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battula, V.L.; Chen, Y.; Cabreira Mda, G.; Ruvolo, V.; Wang, Z.; Ma, W.; Konoplev, S.; Shpall, E.; Lyons, K.; Strunk, D.; et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood 2013, 122, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabe, Y.; Saitoh, K.; Yang, H.; Sekihara, K.; Yamatani, K.; Ruvolo, V.; Taka, H.; Kaga, N.; Kikkawa, M.; Arai, H.; et al. Inhibition of FAO in AML co-cultured with BM adipocytes: Mechanisms of survival and chemosensitization to cytarabine. Sci. Rep. 2018, 8, 16837. [Google Scholar] [CrossRef] [PubMed]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.A.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009, 69, 7867–7874. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e5. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Baccelli, I.; Gareau, Y.; Lehnertz, B.; Gingras, S.; Spinella, J.F.; Corneau, S.; Mayotte, N.; Girard, S.; Frechette, M.; Blouin-Chagnon, V.; et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 2019, 36, 84–99.e8. [Google Scholar] [CrossRef]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef] [Green Version]
- Boultwood, J.; Fidler, C.; Mills, K.I.; Frodsham, P.M.; Kusec, R.; Gaiger, A.; Gale, R.E.; Linch, D.C.; Littlewood, T.J.; Moss, P.A.; et al. Amplification of mitochondrial DNA in acute myeloid leukaemia. Br. J. Haematol. 1996, 95, 426–431. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Raso-Barnett, L.; Scott, M.A.; Ingham, C.J.; Collins, A.; Bowles, K.M.; Rushworth, S.A. PGC-1alpha driven mitochondrial biogenesis in stromal cells underpins mitochondrial trafficking to leukemic blasts. Leukemia 2018, 32, 2073–2077. [Google Scholar] [CrossRef]
- Leverson, J.D.; Cojocari, D. Hematologic Tumor Cell Resistance to the BCL-2 Inhibitor Venetoclax: A Product of Its Microenvironment? Front. Oncol. 2018, 8, 458. [Google Scholar] [CrossRef] [Green Version]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Pratz, K.; Letai, A.; Jonas, B.A.; Wei, A.H.; Pullarkat, V.; Konopleva, M.; Thirman, M.J.; Arellano, M.; Becker, P.S.; et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am. J. Hematol. 2021, 96, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Forte, D.; García-Fernández, M.; Sánchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martín-Pérez, D.; López, J.A.; Costa, A.S.H.; Tronci, L.; Nikitopoulou, E.; et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab. 2020, 32, 829–843.e9. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lei, W.; Chen, X.; Wang, S.; Qian, W. Oxidative stress response induced by chemotherapy in leukemia treatment. Mol. Clin. Oncol. 2018, 8, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, H.; Wang, K. Reactive oxygen species in eradicating acute myeloid leukemic stem cells. Stem Cell Investig. 2014, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Boutin, L.; Arnautou, P.; Trignol, A.; Segot, A.; Farge, T.; Desterke, C.; Soave, S.; Clay, D.; Raffoux, E.; Sarry, J.E.; et al. Mesenchymal stromal cells confer chemoresistance to myeloid leukemia blasts through Side Population functionality and ABC transporter activation. Haematologica 2020, 105, 987–9998. [Google Scholar] [CrossRef]
- Vignon, C.; Debeissat, C.; Bourgeais, J.; Gallay, N.; Kouzi, F.; Anginot, A.; Picou, F.; Guardiola, P.; Ducrocq, E.; Foucault, A.; et al. Involvement of GPx-3 in the Reciprocal Control of Redox Metabolism in the Leukemic Niche. Int. J. Mol. Sci. 2020, 21, 8584. [Google Scholar] [CrossRef]
- Taniguchi Ishikawa, E.; Gonzalez-Nieto, D.; Ghiaur, G.; Dunn, S.K.; Ficker, A.M.; Murali, B.; Madhu, M.; Gutstein, D.E.; Fishman, G.I.; Barrio, L.C.; et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9071–9076. [Google Scholar] [CrossRef] [Green Version]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Austin, R.; Smyth, M.J.; Lane, S.W. Harnessing the immune system in acute myeloid leukaemia. Crit. Rev. Oncol. Hematol. 2016, 103, 62–77. [Google Scholar] [CrossRef] [Green Version]
- Vago, L.; Perna, S.K.; Zanussi, M.; Mazzi, B.; Barlassina, C.; Stanghellini, M.T.; Perrelli, N.F.; Cosentino, C.; Torri, F.; Angius, A.; et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N. Engl. J. Med. 2009, 361, 478–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Le Dieu, R.; Taussig, D.C.; Ramsay, A.G.; Mitter, R.; Miraki-Moud, F.; Fatah, R.; Lee, A.M.; Lister, T.A.; Gribben, J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood 2009, 114, 3909–3916. [Google Scholar] [CrossRef] [Green Version]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Taghiloo, S.; Asgarian-Omran, H. Immune evasion mechanisms in acute myeloid leukemia: A focus on immune checkpoint pathways. Crit. Rev. Oncol. Hematol. 2021, 157, 103164. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2021. [Google Scholar] [CrossRef]
- Coles, S.J.; Hills, R.K.; Wang, E.C.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia 2012, 26, 2146–2148. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Trabanelli, S.; Onofri, C.; Aluigi, M.; Salvestrini, V.; Ocadlikova, D.; Evangelisti, C.; Rutella, S.; De Cristofaro, R.; Ottaviani, E.; et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica 2010, 95, 2022–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovatti, P.E.; Gambacorta, V.; Lorentino, F.; Ciceri, F.; Vago, L. Mechanisms of Leukemia Immune Evasion and Their Role in Relapse after Haploidentical Hematopoietic Cell Transplantation. Front. Immunol. 2020, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Benites, B.D.; da Silva Santos Duarte, A.; Longhini, A.L.F.; Santos, I.; Alvarez, M.C.; de Morais Ribeiro, L.N.; Paula, E.; Saad, S.T.O. Exosomes in the serum of Acute Myeloid Leukemia patients induce dendritic cell tolerance: Implications for immunotherapy. Vaccine 2019, 37, 1377–1383. [Google Scholar] [CrossRef]
- Hong, C.S.; Sharma, P.; Yerneni, S.S.; Simms, P.; Jackson, E.K.; Whiteside, T.L.; Boyiadzis, M. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci. Rep. 2017, 7, 14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hönes, J.M.; Lams, R.F.; Thivakaran, A.; Schütte, J.; Köster, R.; Lennartz, K.; Schroeder, T.; et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a Growth factor independence 1 dependent manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef] [Green Version]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef]
- Chamuleau, M.E.; van de Loosdrecht, A.A.; Hess, C.J.; Janssen, J.J.; Zevenbergen, A.; Delwel, R.; Valk, P.J.; Lowenberg, B.; Ossenkoppele, G.J. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. [Google Scholar] [CrossRef]
- Ragaini, S.; Wagner, S.; Marconi, G.; Parisi, S.; Sartor, C.; Nanni, J.; Cristiano, G.; Talami, A.; Olivi, M.; Ocadlikova, D.; et al. An IDO1-related immune gene signature predicts overall survival in acute myeloid leukemia. Blood Adv. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, R.; Salinas-Illarena, A.; Baldauf, H.M. New strategies to treat AML: Novel insights into AML survival pathways and combination therapies. Leukemia 2021, 35, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.; Salvestrini, V.; Ciciarello, M.; Loscocco, F.; Visani, G.; Parisi, S.; Lecciso, M.; Ocadlikova, D.; Rossi, L.; Gabucci, E.; et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev. Hematol. 2014, 7, 807–818. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, e120974. [Google Scholar] [CrossRef] [Green Version]
- Lamble, A.J.; Lind, E.F. Targeting the Immune Microenvironment in Acute Myeloid Leukemia: A Focus on T Cell Immunity. Front. Oncol. 2018, 8, 213. [Google Scholar] [CrossRef]
- Tajima, F.; Kawatani, T.; Endo, A.; Kawasaki, H. Natural killer cell activity and cytokine production as prognostic factors in adult acute leukemia. Leukemia 1996, 10, 478–482. [Google Scholar] [PubMed]
- Lion, E.; Willemen, Y.; Berneman, Z.N.; Van Tendeloo, V.F.; Smits, E.L. Natural killer cell immune escape in acute myeloid leukemia. Leukemia 2012, 26, 2019–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Brück, O.; Dufva, O.; Hohtari, H.; Blom, S.; Turkki, R.; Ilander, M.; Kovanen, P.; Pallaud, C.; Ramos, P.M.; Lähteenmäki, H.; et al. Immune profiles in acute myeloid leukemia bone marrow associate with patient age, T-cell receptor clonality, and survival. Blood Adv. 2020, 4, 274–286. [Google Scholar] [CrossRef]
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where We Are and What Comes Next? Front. Immunol. 2017, 8, 1578. [Google Scholar] [CrossRef]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Fisher, S.A.; Lamikanra, A.; Dorée, C.; Gration, B.; Tsang, P.; Danby, R.D.; Roberts, D.J. Increased regulatory T cell graft content is associated with improved outcome in haematopoietic stem cell transplantation: A systematic review. Br. J. Haematol. 2017, 176, 448–463. [Google Scholar] [CrossRef] [Green Version]
- Dwarakanath, B.S.; Farooque, A.; Gupta, S. Targeting regulatory T cells for improving cancer therapy: Challenges and prospects. Cancer Rep. (Hoboken) 2018, 1, e21105. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Choudhury, A.; Lladser, A.; Kiessling, R.; Johansson, C.C. Regulatory T cells in cancer. Adv. Cancer Res. 2010, 107, 57–117. [Google Scholar] [CrossRef] [PubMed]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Sopper, S.; Pircher, A.; Gastl, G.; Wolf, A.M. Treg(s) in Cancer: Friends or Foe? J. Cell Physiol. 2015, 230, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Muthu Raja, K.R.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T regulatory cells are associated with adverse clinical features and predict progression in multiple myeloma. PLoS ONE 2012, 7, e47077. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, T.; Lejeune, M.; Salvadó, M.T.; Bosch, R.; García, J.F.; Jaén, J.; Banham, A.H.; Roncador, G.; Montalbán, C.; Piris, M.A. Outcome in Hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin. Cancer Res. 2005, 11, 1467–1473. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Wu, S.Y.; Kang, Y.W.; Lin, K.P.; Chen, T.Y.; Medeiros, L.J.; Chang, K.C. High levels of regulatory T cells in blood are a poor prognostic factor in patients with diffuse large B-cell lymphoma. Am. J. Clin. Pathol. 2015, 144, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Zhang, C.; Xu, Y.; Wang, M.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Mi, Y.; Wang, Y.; et al. Hyperfunction of CD4 CD25 regulatory T cells in de novo acute myeloid leukemia. BMC Cancer 2020, 20, 472. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Czystowska, M.; Mandapathil, M.; Strauss, L.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin. Cancer Res. 2009, 15, 3325–3332. [Google Scholar] [CrossRef] [Green Version]
- Shenghui, Z.; Yixiang, H.; Jianbo, W.; Kang, Y.; Laixi, B.; Yan, Z.; Xi, X. Elevated frequencies of CD4⁺ CD25⁺ CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int. J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef]
- Yang, W.; Xu, Y. Clinical significance of Treg cell frequency in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Bucher, C.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Munn, D.H.; Levine, B.L.; Riddle, M.; June, C.H.; Vallera, D.A.; et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood 2009, 114, 3793–3802. [Google Scholar] [CrossRef]
- Wang, R.; Feng, W.; Wang, H.; Wang, L.; Yang, X.; Yang, F.; Zhang, Y.; Liu, X.; Zhang, D.; Ren, Q.; et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett. 2020, 469, 151–161. [Google Scholar] [CrossRef]
- Nishikawa, H. Regulatory T cells in cancer immunotherapy. Rinsho Ketsueki 2014, 55, 2183–2189. [Google Scholar] [CrossRef] [Green Version]
- Saida, Y.; Watanabe, S.; Tanaka, T.; Baba, J.; Sato, K.; Shoji, S.; Igarashi, N.; Kondo, R.; Okajima, M.; Koshio, J.; et al. Critical Roles of Chemoresistant Effector and Regulatory T Cells in Antitumor Immunity after Lymphodepleting Chemotherapy. J. Immunol. 2015, 195, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valzasina, B.; Piconese, S.; Guiducci, C.; Colombo, M.P. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25- lymphocytes is thymus and proliferation independent. Cancer Res. 2006, 66, 4488–4495. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Workman, C.J.; Vignali, D.A. Targeting regulatory T cells in tumors. FEBS J. 2016, 283, 2731–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hersh, E.M.; Whitecar, J.P.; McCredie, K.B.; Bodey, G.P.; Freireich, E.J. Chemotherapy, immunocompetence, immunosuppression and prognosis in acute leukemia. N. Engl. J. Med. 1971, 285, 1211–1216. [Google Scholar] [CrossRef]
- Hersh, E.M.; Gutterman, J.U.; Mavligit, G.M.; McCredie, K.B.; Burgess, M.A.; Matthews, A.; Freireich, E.J. Serial studies of immunocompetence of patients undergoing chemotherapy for acute leukemia. J. Clin. Investig. 1974, 54, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Wu, J.; Li, C.G.; Jiang, H.W.; Xu, M.; Du, M.; Yin, Z.; Mei, H.; Hu, Y. Characterization of Immune Dysfunction and Identification of Prognostic Immune-Related Risk Factors in Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 1763–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Bu, J.; Zhou, M.; Sido, J.; Lin, Y.; Liu, G.; Lin, Q.; Xu, X.; Leavenworth, J.W.; Shen, E. CD8+ T cells expressing both PD-1 and TIGIT but not CD226 are dysfunctional in acute myeloid leukemia (AML) patients. Clin. Immunol. 2018, 190, 64–73. [Google Scholar] [CrossRef]
- Behl, D.; Porrata, L.F.; Markovic, S.N.; Letendre, L.; Pruthi, R.K.; Hook, C.C.; Tefferi, A.; Elliot, M.A.; Kaufmann, S.H.; Mesa, R.A.; et al. Absolute lymphocyte count recovery after induction chemotherapy predicts superior survival in acute myelogenous leukemia. Leukemia 2006, 20, 29–34. [Google Scholar] [CrossRef]
- Malkan, U.Y.; Gunes, G.; Isik, A.; Eliacik, E.; Etgul, S.; Aslan, T.; Balaban, M.S.; Haznedaroglu, I.C.; Demiroglu, H.; Goker, H.; et al. Rebound Thrombocytosis following Induction Chemotherapy is an Independent Predictor of a Good Prognosis in Acute Myeloid Leukemia Patients Attaining First Complete Remission. Acta Haematol. 2015, 134, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Yanada, M.; Borthakur, G.; Garcia-Manero, G.; Ravandi, F.; Faderl, S.; Pierce, S.; Kantarjian, H.; Estey, E. Blood counts at time of complete remission provide additional independent prognostic information in acute myeloid leukemia. Leuk. Res. 2008, 32, 1505–1509. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.I.; Trepel, M.; Kunzmann, R.; Lais, A.; Engelhardt, R.; Lübbert, M. Hematologic and molecular spontaneous remission following sepsis in acute monoblastic leukemia with translocation (9;11): A case report and review of the literature. Eur. J. Haematol. 2004, 73, 62–66. [Google Scholar] [CrossRef]
- Markey, K.A.; MacDonald, K.P.; Hill, G.R. The biology of graft-versus-host disease: Experimental systems instructing clinical practice. Blood 2014, 124, 354–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol. 2021, 11, 656218. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020, 12, eaaz0463. [Google Scholar] [CrossRef]
- Salik, B.; Smyth, M.J.; Nakamura, K. Targeting immune checkpoints in hematological malignancies. J. Hematol. Oncol. 2020, 13, 111. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Mandero, G.; Konopleva, M.Y.; Alfayez, M.; Pemmaraju, N.; Kadia, T.M.; DiNardo, C.; Cortes, J.; Ravandi, F.; Abbas, H.; et al. Azacitidine (AZA) with Nivolumab (Nivo), and AZA with Nivo + Ipilimumab (Ipi) in relapsed/refractory acute myeloid leukemia: A non-randomized, prospective, phase 2 study. Blood 2019, 134 (Suppl. 1), 830. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [Green Version]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef]
- Curti, A.; Trabanelli, S.; Salvestrini, V.; Baccarani, M.; Lemoli, R.M. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: Focus on hematology. Blood 2009, 113, 2394–2401. [Google Scholar] [CrossRef]
- Curti, A.; Aluigi, M.; Pandolfi, S.; Ferri, E.; Isidori, A.; Salvestrini, V.; Durelli, I.; Horenstein, A.L.; Fiore, F.; Massaia, M.; et al. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia 2007, 21, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhuansun, Y.; Chen, R.; Li, J.; Ran, P. Immunomodulation of mesenchymal stromal cells on regulatory T cells and its possible mechanism. Exp. Cell Res. 2014, 324, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Jiang, J.; Arp, J.; Liu, W.; Garcia, B.; Wang, H. Regulatory T-cell generation and kidney allograft tolerance induced by mesenchymal stem cells associated with indoleamine 2,3-dioxygenase expression. Transplantation 2010, 90, 1312–1320. [Google Scholar] [CrossRef]
- He, Y.; Zhou, S.; Liu, H.; Shen, B.; Zhao, H.; Peng, K.; Wu, X. Indoleamine 2,3-Dioxgenase Transfected Mesenchymal Stem Cells Induce Kidney Allograft Tolerance by Increasing the Production and Function of Regulatory T Cells. Transplantation 2015, 99, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Mansour, I.; Zayed, R.A.; Said, F.; Latif, L.A. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology 2016, 21, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Ghannam, S.; Bouffi, C.; Djouad, F.; Jorgensen, C.; Noel, D. Immunosuppression by mesenchymal stem cells: Mechanisms and clinical applications. Stem Cell Res. Ther. 2010, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Jafarzadeh, N.; Safari, Z.; Pornour, M.; Amirizadeh, N.; Forouzandeh Moghadam, M.; Sadeghizadeh, M. Alteration of cellular and immune-related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J. Cell Physiol. 2019, 234, 3697–3710. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallipoli, P.; Pellicano, F.; Morrison, H.; Laidlaw, K.; Allan, E.K.; Bhatia, R.; Copland, M.; Jorgensen, H.G.; Holyoake, T.L. Autocrine TNF-alpha production supports CML stem and progenitor cell survival and enhances their proliferation. Blood 2013, 122, 3335–3339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Z.; Zhou, J. Tumor necrosis factor alpha in the onset and progression of leukemia. Exp. Hematol. 2017, 45, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castaño, J.; Gómez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep. 2017, 8, 1573–1586. [Google Scholar] [CrossRef] [Green Version]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- Gordon, P.M.; Dias, S.; Williams, D.A. Cytokines secreted by bone marrow stromal cells protect c-KIT mutant AML cells from c-KIT inhibitor-induced apoptosis. Leukemia 2014, 28, 2257–2260. [Google Scholar] [CrossRef] [Green Version]
- Lecciso, M.; Ocadlikova, D.; Sangaletti, S.; Trabanelli, S.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Portararo, P.; Jandus, C.; Bontadini, A.; et al. ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front. Immunol. 2017, 8, 1918. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciciarello, M.; Corradi, G.; Forte, D.; Cavo, M.; Curti, A. Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance? Cancers 2021, 13, 5319. https://doi.org/10.3390/cancers13215319
Ciciarello M, Corradi G, Forte D, Cavo M, Curti A. Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance? Cancers. 2021; 13(21):5319. https://doi.org/10.3390/cancers13215319
Chicago/Turabian StyleCiciarello, Marilena, Giulia Corradi, Dorian Forte, Michele Cavo, and Antonio Curti. 2021. "Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance?" Cancers 13, no. 21: 5319. https://doi.org/10.3390/cancers13215319