
PARP Inhibitors and Haematological Malignancies—Friend or Foe?


Abstract
:Simple Summary
Abstract

1. Introduction
2. PARP Structure and Function
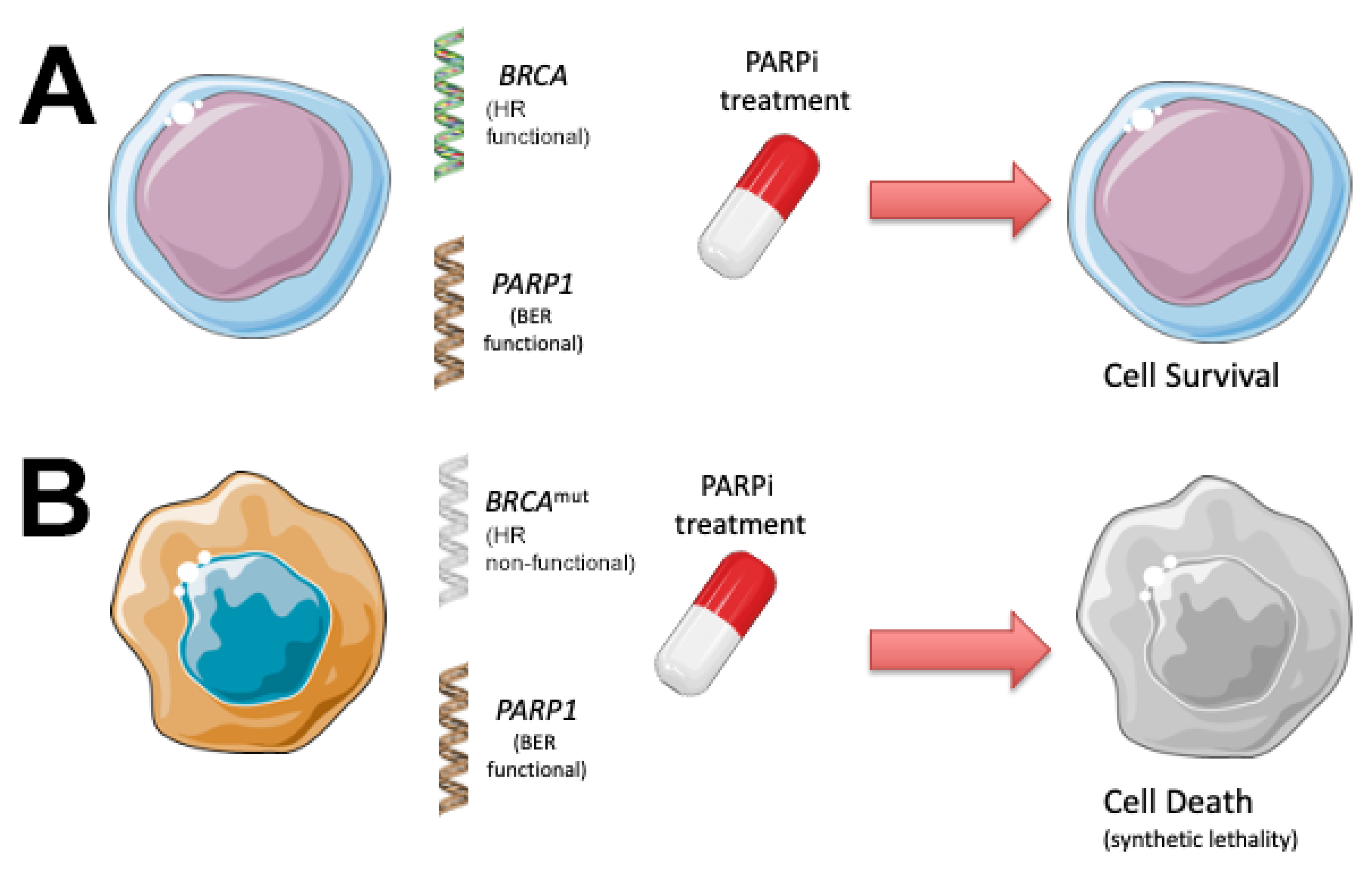
3. PARP Inhibitors Use Synthetic Lethality to Kill Cancer Cells with Defects in DNA Repair
4. PARP Inhibitors as a Potential Treatment for Haematological Malignancies
4.1. Acute Leukaemias
4.1.1. Acute Lymphoblastic Leukaemia
4.1.2. Acute Myeloid Leukaemia
4.2. Chronic Leukaemias
4.2.1. Chronic Lymphocytic Leukaemia
4.2.2. Chronic Myeloid Leukaemia
4.3. Lymphomas
4.3.1. Non-Hodgkin Lymphoma
4.3.2. Cutaneous Lymphoma
4.3.3. Clinical Trials Examining PARPi in Lymphomas
4.4. Multiple Myeloma
4.5. Myeloproliferative Neoplasms
5. Treatment of Cancer Patients with PARPi Has Been Associated with Increased Risk of Haematological Toxicities
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhmoud, J.F.; Mustafa, A.G.; Malki, M.I. Targeting DNA Repair Pathways in Hematological Malignancies. Int. J. Mol. Sci. 2020, 21, 7365. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Martin, N.M.; Smith, G.C. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene 2007, 26, 7816–7824. [Google Scholar] [CrossRef] [Green Version]
- Morice, P.M.; Leary, A.; Dolladille, C.; Chretien, B.; Poulain, L.; Gonzalez-Martin, A.; Moore, K.; O’Reilly, E.M.; Ray-Coquard, I.; Alexandre, J. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: A safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021, 8, e122–e134. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and its associated nucleases in DNA damage response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.O.; Khan, F.A.; Galindo-Campos, M.A.; Yelamos, J. Understanding specific functions of PARP-2: New lessons for cancer therapy. Am. J. Cancer Res. 2016, 6, 1842–1863. [Google Scholar] [PubMed]
- Sodhi, R.K.; Singh, N.; Jaggi, A.S. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul. Pharmacol. 2010, 53, 77–87. [Google Scholar] [CrossRef]
- Huber, A.; Bai, P.; de Murcia, J.M.; de Murcia, G. PARP-1, PARP-2 and ATM in the DNA damage response: Functional synergy in mouse development. DNA Repair 2004, 3, 1103–1108. [Google Scholar] [CrossRef]
- Kumar, M.; Jaiswal, R.K.; Yadava, P.K.; Singh, R.P. An assessment of poly (ADP-ribose) polymerase-1 role in normal and cancer cells. Biofactors 2020, 46, 894–905. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [Green Version]
- Pearsall, E.A.; Lincz, L.F.; Skelding, K.A. The Role of DNA Repair Pathways in AML Chemosensitivity. Curr. Drug Targets 2018, 19, 1205–1219. [Google Scholar] [CrossRef]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, M.; Rosen, D.; Fields, S.; Cesano, A.; Budman, D.R. Poly(ADP-ribose) polymerase-1 inhibition: Preclinical and clinical development of synthetic lethality. Mol. Med. 2011, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmana, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraris, D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef]
- Steffen, J.D.; Brody, J.R.; Armen, R.S.; Pascal, J.M. Structural Implications for Selective Targeting of PARPs. Front. Oncol. 2013, 3, 301. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, A.J. The potential role and application of PARP inhibitors in cancer treatment. Br. Med. Bull. 2009, 89, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Woon, E.C.; Threadgill, M.D. Poly(ADP-ribose)polymerase inhibition—Where now? Curr. Med. Chem. 2005, 12, 2373–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underhill, C.; Toulmonde, M.; Bonnefoi, H. A review of PARP inhibitors: From bench to bedside. Ann. Oncol. 2011, 22, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Liscio, P.; Camaioni, E.; Carotti, A.; Pellicciari, R.; Macchiarulo, A. From polypharmacology to target specificity: The case of PARP inhibitors. Curr. Top. Med. Chem. 2013, 13, 2939–2954. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Sandhu, S.K.; de Bono, J.S. PARP inhibitors: Mechanism of action and their potential role in the prevention and treatment of cancer. Drugs 2012, 72, 1579–1590. [Google Scholar] [CrossRef]
- van Wietmarschen, N.; Nussenzweig, A. Mechanism for Synthetic Lethality in BRCA-Deficient Cancers: No Longer Lagging Behind. Mol. Cell 2018, 71, 877–878. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Nagel, R.; Semenova, E.A.; Berns, A. Drugging the addict: Non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. [Google Scholar] [CrossRef]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5791. [Google Scholar] [CrossRef] [Green Version]
- George, A.; Banerjee, S.; Kaye, S. Olaparib and somatic BRCA mutations. Oncotarget 2017, 8, 43598–43599. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, G.P.; Degar, J.; Hinze, L.; Kochupurakkal, B.; Vo, C.D.; Burns, M.A.; Moreau, L.; Ganesa, C.; Roderick, J.; Peirs, S.; et al. Fanconi-BRCA pathway mutations in childhood T-cell acute lymphoblastic leukemia. PLoS ONE 2019, 14, e0221288. [Google Scholar] [CrossRef] [PubMed]
- Goricar, K.; Erculj, N.; Faganel Kotnik, B.; Debeljak, M.; Hovnik, T.; Jazbec, J.; Dolzan, V. The association of folate pathway and DNA repair polymorphisms with susceptibility to childhood acute lymphoblastic leukemia. Gene 2015, 562, 203–209. [Google Scholar] [CrossRef]
- Piao, J.; Takai, S.; Kamiya, T.; Inukai, T.; Sugita, K.; Ohyashiki, K.; Delia, D.; Masutani, M.; Mizutani, S.; Takagi, M. Poly (ADP-ribose) polymerase inhibitors selectively induce cytotoxicity in TCF3-HLF-positive leukemic cells. Cancer Lett. 2017, 386, 131–140. [Google Scholar] [CrossRef]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; Te Kronnie, G.; Meyer, L.H.; Mohr, F.; et al. TET1 promotes growth of T-cell acute lymphoblastic leukemia and can be antagonized via PARP inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef]
- Bai, X.T.; Moles, R.; Chaib-Mezrag, H.; Nicot, C. Small PARP inhibitor PJ-34 induces cell cycle arrest and apoptosis of adult T-cell leukemia cells. J. Hematol. Oncol. 2015, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Jasek, E.; Gajda, M.; Lis, G.J.; Jasinska, M.; Litwin, J.A. Combinatorial effects of PARP inhibitor PJ34 and histone deacetylase inhibitor vorinostat on leukemia cell lines. Anticancer Res. 2014, 34, 1849–1856. [Google Scholar] [PubMed]
- Horvat, L.; Antica, M.; Matulic, M. Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells. Cells 2018, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, T.M.; Jenkins, G.; Pati, D.; Zhang, L.; Dolan, M.E.; Ribes-Zamora, A.; Bertuch, A.A.; Blaney, S.M.; Delaney, S.L.; Hegde, M.; et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: Influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol. Cancer Ther. 2009, 8, 2232–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzacappa, M.V.; Ronchini, C.; Faretta, M.; Iacobucci, I.; Di Rora, A.G.; Martinelli, G.; Meyer, L.H.; Debatin, K.M.; Orecchioni, S.; Bertolini, F.; et al. The Combination of the PARP Inhibitor Rucaparib and 5FU Is an Effective Strategy for Treating Acute Leukemias. Mol. Cancer Ther. 2015, 14, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cai, W.; Zhang, W.; Chen, X.; Dong, W.; Tang, D.; Zhang, Y.; Ji, C.; Zhang, M. Inhibition of poly(ADP-ribose) polymerase 1 protects against acute myeloid leukemia by suppressing the myeloproliferative leukemia virus oncogene. Oncotarget 2015, 6, 27490–27504. [Google Scholar] [CrossRef] [Green Version]
- Gaymes, T.J.; Shall, S.; MacPherson, L.J.; Twine, N.A.; Lea, N.C.; Farzaneh, F.; Mufti, G.J. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: Potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 2009, 94, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepnik, M.; Spryszynska, S.; Gorzkiewicz, A.; Ferlinska, M. Cytotoxicity of anticancer drugs and PJ-34 (poly(ADP-ribose)polymerase-1 (PARP-1) inhibitor) on HL-60 and Jurkat cells. Adv. Clin. Exp. Med. 2017, 26, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. BRCA1, PARP1 and gammaH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim. Biophys. Acta 2015, 1852, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Uzui, K.; Nishi, R.; Shigemi, H.; Ueda, T. Gemtuzumab ozogamicin and olaparib exert synergistic cytotoxicity in CD33-positive HL-60 myeloid leukemia cells. Anticancer Res. 2014, 34, 5487–5494. [Google Scholar] [PubMed]
- Orta, M.L.; Hoglund, A.; Calderon-Montano, J.M.; Dominguez, I.; Burgos-Moron, E.; Visnes, T.; Pastor, N.; Strom, C.; Lopez-lazaro, M.; Helleday, T. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2’-deoxycytidine lesions. Nucleic Acids Res. 2014, 42, 9108–9120. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef] [Green Version]
- Nieborowska-Skorska, M.; Paietta, E.M.; Levine, R.L.; Fernandez, H.F.; Tallman, M.S.; Litzow, M.R.; Skorski, T. Inhibition of the mutated c-KIT kinase in AML1-ETO-positive leukemia cells restores sensitivity to PARP inhibitor. Blood Adv. 2019, 3, 4050–4054. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.B.; Snedeker, J.C.; Baturin, D.; Gardner, L.; Fosmire, S.P.; Zhou, C.; Jordan, C.T.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. A Small-Molecule Inhibitor of WEE1, AZD1775, Synergizes with Olaparib by Impairing Homologous Recombination and Enhancing DNA Damage and Apoptosis in Acute Leukemia. Mol. Cancer Ther. 2017, 16, 2058–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2018, 9, 3908–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giansanti, M.; De Gabrieli, A.; Prete, S.P.; Ottone, T.; Divona, M.D.; Karimi, T.; Ciccarone, F.; Voso, M.T.; Graziani, G.; Faraoni, I. Poly(ADP-Ribose) Polymerase Inhibitors for Arsenic Trioxide-Resistant Acute Promyelocytic Leukemia: Synergistic In Vitro Antitumor Effects with Hypomethylating Agents or High-Dose Vitamin C. J. Pharmacol. Exp. Ther. 2021, 377, 385–397. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Gaymes, T.J.; Mohamedali, A.M.; Patterson, M.; Matto, N.; Smith, A.; Kulasekararaj, A.; Chelliah, R.; Curtin, N.; Farzaneh, F.; Shall, S.; et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica 2013, 98, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Sullivan, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Hoser, G.; Maifrede, S.; Martinez, E.; Di Marcantonio, D.; Bolton-Gillespie, E.; Cramer-Morales, K.; et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J. Clin. Investig. 2017, 127, 2392–2406. [Google Scholar] [CrossRef]
- Le, B.V.; Podszywalow-Bartnicka, P.; Maifrede, S.; Sullivan-Reed, K.; Nieborowska-Skorska, M.; Golovine, K.; Yao, J.C.; Nejati, R.; Cai, K.Q.; Caruso, L.B.; et al. TGFbetaR-SMAD3 Signaling Induces Resistance to PARP Inhibitors in the Bone Marrow Microenvironment. Cell Rep. 2020, 33, 108221. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Radivoyevitch, T.; Nagata, Y.; Khurshed, M.; Przychodzen, B.; Makishima, H.; Xu, M.; Bleeker, F.E.; Wilmink, J.W.; Carraway, H.E.; et al. IDH1/2 Mutations Sensitize Acute Myeloid Leukemia to PARP Inhibition and This Is Reversed by IDH1/2-Mutant Inhibitors. Clin. Cancer Res. 2018, 24, 1705–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Willmore, E.; Austin, C.A.; Curtin, N.J. The novel poly(ADP-Ribose) polymerase inhibitor, AG14361, sensitizes cells to topoisomerase I poisons by increasing the persistence of DNA strand breaks. Clin. Cancer Res. 2005, 11, 8449–8457. [Google Scholar] [CrossRef] [Green Version]
- Tobin, L.A.; Robert, C.; Rapoport, A.P.; Gojo, I.; Baer, M.R.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene 2013, 32, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Nieborowska-Skorska, M.; Maifrede, S.; Ye, M.; Toma, M.; Hewlett, E.; Gordon, J.; Le, B.V.; Sliwinski, T.; Zhao, H.; Piwocka, K.; et al. Non-NAD-like PARP1 inhibitor enhanced synthetic lethal effect of NAD-like PARP inhibitors against BRCA1-deficient leukemia. Leuk. Lymphoma 2019, 60, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.Y.; Kan, W.M. Poly ADP-ribose polymerase inhibition suppresses cisplatin toxicity in chronic myeloid leukemia cells. Anticancer Drugs 2017, 28, 316–321. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Maifrede, S.; Le, B.V.; Nieborowska-Skorska, M.; Piwocka, K.; Skorski, T. PARP1 inhibitor eliminated imatinib-refractory chronic myeloid leukemia cells in bone marrow microenvironment conditions. Leuk. Lymphoma 2019, 60, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, H.; Song, H.; Feng, X.; Zhou, C.; Huo, Z. Targeting autophagy potentiates the anti-tumor effect of PARP inhibitor in pediatric chronic myeloid leukemia. AMB Express 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Dilley, R.L.; Poh, W.; Gladstone, D.E.; Herman, J.G.; Showel, M.M.; Karp, J.E.; McDevitt, M.A.; Pratz, K.W. Poly(ADP-ribose) polymerase inhibitor CEP-8983 synergizes with bendamustine in chronic lymphocytic leukemia cells in vitro. Leuk. Res 2014, 38, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef] [Green Version]
- Herriott, A.; Tudhope, S.J.; Junge, G.; Rodrigues, N.; Patterson, M.J.; Woodhouse, L.; Lunec, J.; Hunter, J.E.; Mulligan, E.A.; Cole, M.; et al. PARP1 expression, activity and ex vivo sensitivity to the PARP inhibitor, talazoparib (BMN 673), in chronic lymphocytic leukaemia. Oncotarget 2015, 6, 43978–43991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golla, R.M.; Li, M.; Shen, Y.; Ji, M.; Yan, Y.; Fu, K.; Greiner, T.C.; McKeithan, T.W.; Chan, W.C. Inhibition of poly(ADP-ribose) polymerase (PARP) and ataxia telangiectasia mutated (ATM) on the chemosensitivity of mantle cell lymphoma to agents that induce DNA strand breaks. Hematol. Oncol. 2012, 30, 175–179. [Google Scholar] [CrossRef]
- Williamson, C.T.; Muzik, H.; Turhan, A.G.; Zamo, A.; O’Connor, M.J.; Bebb, D.G.; Lees-Miller, S.P. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 2010, 9, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Maifrede, S.; Martin, K.; Podszywalow-Bartnicka, P.; Sullivan-Reed, K.; Langer, S.K.; Nejati, R.; Dasgupta, Y.; Hulse, M.; Gritsyuk, D.; Nieborowska-Skorska, M.; et al. IGH/MYC Translocation Associates with BRCA2 Deficiency and Synthetic Lethality to PARP1 Inhibitors. Mol. Cancer Res. 2017, 15, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, N.G.; James, E.; Wahl, R.L. Poly(ADP-ribose) polymerase inhibitors combined with external beam and radioimmunotherapy to treat aggressive lymphoma. Nucl. Med. Commun. 2011, 32, 1046–1051. [Google Scholar] [CrossRef] [Green Version]
- Parvin, S.; Ramirez-Labrada, A.; Aumann, S.; Lu, X.; Weich, N.; Santiago, G.; Cortizas, E.M.; Sharabi, E.; Zhang, Y.; Sanchez-Garcia, I.; et al. LMO2 Confers Synthetic Lethality to PARP Inhibition in DLBCL. Cancer Cell 2019, 36, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.; Rueter, J.; Rajan, S.; Zhang, R.; Shopland, L. Additive and synergistic inhibition of mantle cell lymphoma cell growth by combining olaparib with ibrutinib. J. Cell Biochem. 2018, 119, 5843–5851. [Google Scholar] [CrossRef] [PubMed]
- Lemchak, D.; Banerjee, S.; Digambar, S.S.; Hood, B.L.; Conrads, T.P.; Jedrych, J.; Geskin, L.; Akilov, O.E. Therapeutic and prognostic significance of PARP-1 in advanced mycosis fungoides and Sezary syndrome. Exp. Dermatol. 2018, 27, 188–190. [Google Scholar] [CrossRef]
- Weltin, D.; Holl, V.; Hyun, J.W.; Dufour, P.; Marchal, J.; Bischoff, P. Effect of 6(5H)-phenanthridinone, a poly (ADP-ribose)polymerase inhibitor, and ionizing radiation on the growth of cultured lymphoma cells. Int. J. Radiat. Biol. 1997, 72, 685–692. [Google Scholar] [CrossRef]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. The PARP inhibitor olaparib enhances the cytotoxicity of combined gemcitabine, busulfan and melphalan in lymphoma cells. Leuk. Lymphoma 2017, 58, 2705–2716. [Google Scholar] [CrossRef]
- Kruglov, O.; Wu, X.; Hwang, S.T.; Akilov, O.E. The synergistic proapoptotic effect of PARP-1 and HDAC inhibition in cutaneous T-cell lymphoma is mediated via Blimp-1. Blood Adv. 2020, 4, 4788–4797. [Google Scholar] [CrossRef] [PubMed]
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 helicase is involved in replication stress survival and drug resistance in multiple myeloma. Leukemia 2017, 31, 2104–2113. [Google Scholar] [CrossRef] [Green Version]
- Xiong, T.; Wei, H.; Chen, X.; Xiao, H. PJ34, a poly(ADP-ribose) polymerase (PARP) inhibitor, reverses melphalan-resistance and inhibits repair of DNA double-strand breaks by targeting the FA/BRCA pathway in multidrug resistant multiple myeloma cell line RPMI8226/R. Int. J. Oncol. 2015, 46, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbarulo, A.; Iansante, V.; Chaidos, A.; Naresh, K.; Rahemtulla, A.; Franzoso, G.; Karadimitris, A.; Haskard, D.O.; Papa, S.; Bubici, C. Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene 2013, 32, 4231–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2021, 106, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagpulinsa, D.A.; Ayyadevara, S.; Yaccoby, S.; Shmookler Reis, R.J. A Cyclin-Dependent Kinase Inhibitor, Dinaciclib, Impairs Homologous Recombination and Sensitizes Multiple Myeloma Cells to PARP Inhibition. Mol. Cancer Ther. 2016, 15, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [Green Version]
- Pratz, K.W.; Koh, B.D.; Patel, A.G.; Flatten, K.S.; Poh, W.; Herman, J.G.; Dilley, R.; Harrell, M.I.; Smith, B.D.; Karp, J.E.; et al. Poly (ADP-Ribose) Polymerase Inhibitor Hypersensitivity in Aggressive Myeloproliferative Neoplasms. Clin. Cancer Res. 2016, 22, 3894–3902. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.R.; Senyuk, V.; Rodriguez, N.S.; Oh, A.L.; Bonetti, E.; Mahmud, D.; Barosi, G.; Mahmud, N.; Rondelli, D. Synergistic Cytotoxic Effect of Busulfan and the PARP Inhibitor Veliparib in Myeloproliferative Neoplasms. Biol. Blood Marrow Transplant. 2019, 25, 855–860. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Maifrede, S.; Dasgupta, Y.; Sullivan, K.; Flis, S.; Le, B.V.; Solecka, M.; Belyaeva, E.A.; Kubovcakova, L.; Nawrocki, M.; et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017, 130, 2848–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraoni, I.; Consalvo, M.I.; Aloisio, F.; Fabiani, E.; Giansanti, M.; Di Cristino, F.; Falconi, G.; Tentori, L.; Di Veroli, A.; Curzi, P.; et al. Cytotoxicity and Differentiating Effect of the Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Myelodysplastic Syndromes. Cancers 2019, 11, 1373. [Google Scholar] [CrossRef] [Green Version]
- Tothova, Z.; Valton, A.L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Qu, C.; Varotto, E.; Janke, L.J.; Yang, X.; Seth, A.; Shelat, A.; Friske, J.D.; Fukano, R.; Yu, J.; et al. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood 2021, 137, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Knittel, G.; Rehkamper, T.; Korovkina, D.; Liedgens, P.; Fritz, C.; Torgovnick, A.; Al-Baldawi, Y.; Al-Maarri, M.; Cun, Y.; Fedorchenko, O.; et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat. Commun. 2017, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Leonetti, C.; Scarsella, M.; d’Amati, G.; Portarena, I.; Zupi, G.; Bonmassar, E.; Graziani, G. Combined treatment with temozolomide and poly(ADP-ribose) polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site. Blood 2002, 99, 2241–2244. [Google Scholar] [CrossRef]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef]
- Belgnaoui, S.M.; Fryrear, K.A.; Nyalwidhe, J.O.; Guo, X.; Semmes, O.J. The viral oncoprotein tax sequesters DNA damage response factors by tethering MDC1 to chromatin. J. Biol. Chem. 2010, 285, 32897–32905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef] [Green Version]
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casorelli, I.; Tenedini, E.; Tagliafico, E.; Blasi, M.F.; Giuliani, A.; Crescenzi, M.; Pelosi, E.; Testa, U.; Peschle, C.; Mele, L.; et al. Identification of a molecular signature for leukemic promyelocytes and their normal counterparts: Focus on DNA repair genes. Leukemia 2006, 20, 1978–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Hu, P.; Ye, T.Z.; Stan, R.; Ellis, N.A.; Pandolfi, P.P. A role for PML and the nuclear body in genomic stability. Oncogene 1999, 18, 7941–7947. [Google Scholar] [CrossRef] [Green Version]
- Boichuk, S.; Hu, L.; Makielski, K.; Pandolfi, P.P.; Gjoerup, O.V. Functional connection between Rad51 and PML in homology-directed repair. PLoS ONE 2011, 6, e25814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef]
- Hatano, Y.; Tamada, M.; Matsuo, M.; Hara, A. Molecular Trajectory of BRCA1 and BRCA2 Mutations. Front. Oncol. 2020, 10, 361. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Seedhouse, C.H.; Hunter, H.M.; Lloyd-Lewis, B.; Massip, A.M.; Pallis, M.; Carter, G.I.; Grundy, M.; Shang, S.; Russell, N.H. DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia 2006, 20, 2130–2136. [Google Scholar] [CrossRef]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [Green Version]
- Ghelli Luserna di Rora, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Ha, D.H.; Min, A.; Kim, S.; Jang, H.; Kim, S.H.; Kim, H.J.; Ryu, H.S.; Ku, J.L.; Lee, K.H.; Im, S.A. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci. Rep. 2020, 10, 9930. [Google Scholar] [CrossRef]
- Faurschou, A. Role of tumor necrosis factor-alpha in the regulation of keratinocyte cell cycle and DNA repair after ultraviolet-B radiation. Dan. Med. Bull. 2010, 57, B4179. [Google Scholar]
- Gojo, I.; Beumer, J.H.; Pratz, K.W.; McDevitt, M.A.; Baer, M.R.; Blackford, A.L.; Smith, B.D.; Gore, S.D.; Carraway, H.E.; Showel, M.M.; et al. A Phase 1 Study of the PARP Inhibitor Veliparib in Combination with Temozolomide in Acute Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.X.; Kummar, S.; Steinberg, S.M.; Murgo, A.J.; Gutierrez, M.; Rubinstein, L.; Nguyen, D.; Kaur, G.; Chen, A.P.; Giranda, V.L.; et al. Immunohistochemical detection of poly(ADP-ribose) polymerase inhibition by ABT-888 in patients with refractory solid tumors and lymphomas. Cancer Biol. Ther. 2009, 8, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Kinders, R.; Gutierrez, M.E.; Rubinstein, L.; Parchment, R.E.; Phillips, L.R.; Ji, J.; Monks, A.; Low, J.A.; Chen, A.; et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J. Clin. Oncol. 2009, 27, 2705–2711. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, A.; Ji, J.; Zhang, Y.; Reid, J.M.; Ames, M.; Jia, L.; Weil, M.; Speranza, G.; Murgo, A.J.; et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71, 5626–5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummar, S.; Ji, J.; Morgan, R.; Lenz, H.J.; Puhalla, S.L.; Belani, C.P.; Gandara, D.R.; Allen, D.; Kiesel, B.; Beumer, J.H.; et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 1726–1734. [Google Scholar] [CrossRef] [Green Version]
- Pratz, K.W.; Rudek, M.A.; Gojo, I.; Litzow, M.R.; McDevitt, M.A.; Ji, J.; Karnitz, L.M.; Herman, J.G.; Kinders, R.J.; Smith, B.D.; et al. A Phase I Study of Topotecan, Carboplatin and the PARP Inhibitor Veliparib in Acute Leukemias, Aggressive Myeloproliferative Neoplasms, and Chronic Myelomonocytic Leukemia. Clin. Cancer Res. 2017, 23, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Soumerai, J.D.; Zelenetz, A.D.; Moskowitz, C.H.; Palomba, M.L.; Hamlin, P.A., Jr.; Noy, A.; Straus, D.J.; Moskowitz, A.J.; Younes, A.; Matasar, M.J.; et al. The PARP Inhibitor Veliparib Can Be Safely Added to Bendamustine and Rituximab and Has Preliminary Evidence of Activity in B-Cell Lymphoma. Clin. Cancer Res. 2017, 23, 4119–4126. [Google Scholar] [CrossRef] [Green Version]
- Pratt, G.; Yap, C.; Oldreive, C.; Slade, D.; Bishop, R.; Griffiths, M.; Dyer, M.J.S.; Fegan, C.; Oscier, D.; Pettitt, A.; et al. A multi-centre phase I trial of the PARP inhibitor olaparib in patients with relapsed chronic lymphocytic leukaemia, T-prolymphocytic leukaemia or mantle cell lymphoma. Br. J. Haematol. 2018, 182, 429–433. [Google Scholar] [CrossRef]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martin-Garcia, D.; Bea, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Popp, H.D.; Kohl, V.; Naumann, N.; Flach, J.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Saussele, S.; Hofmann, W.K.; et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Absar, M.; Mahmood, A.; Akhtar, T.; Basit, S.; Ramzan, K.; Jameel, A.; Afzal, S.; Ullah, A.; Qureshi, K.; Alanazi, N.; et al. Whole exome sequencing identifies a novel FANCD2 gene splice site mutation associated with disease progression in chronic myeloid leukemia: Implication in targeted therapy of advanced phase CML. Pak. J. Pharm. Sci. 2020, 33, 1419–1426. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef] [Green Version]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef] [Green Version]
- Carrassa, L.; Colombo, I.; Damia, G.; Bertoni, F. Targeting the DNA damage response for patients with lymphoma: Preclinical and clinical evidences. Cancer Treat. Rev. 2020, 90, 102090. [Google Scholar] [CrossRef]
- De Miranda, N.F.; Peng, R.; Georgiou, K.; Wu, C.; Falk Sorqvist, E.; Berglund, M.; Chen, L.; Gao, Z.; Lagerstedt, K.; Lisboa, S.; et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013, 210, 1729–1742. [Google Scholar] [CrossRef] [PubMed]
- Rendleman, J.; Antipin, Y.; Reva, B.; Adaniel, C.; Przybylo, J.A.; Dutra-Clarke, A.; Hansen, N.; Heguy, A.; Huberman, K.; Borsu, L.; et al. Genetic variation in DNA repair pathways and risk of non-Hodgkin’s lymphoma. PLoS ONE 2014, 9, e101685. [Google Scholar] [CrossRef]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef] [Green Version]
- Monroy, C.M.; Cortes, A.C.; Lopez, M.; Rourke, E.; Etzel, C.J.; Younes, A.; Strom, S.S.; El-Zein, R. Hodgkin lymphoma risk: Role of genetic polymorphisms and gene-gene interactions in DNA repair pathways. Mol. Carcinog. 2011, 50, 825–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkozy, C.; Ribrag, V. Novel agents for mantle cell lymphoma: Molecular rational and clinical data. Expert Opin. Investig. Drugs 2020, 29, 555–566. [Google Scholar] [CrossRef]
- Hill, D.A.; Wang, S.S.; Cerhan, J.R.; Davis, S.; Cozen, W.; Severson, R.K.; Hartge, P.; Wacholder, S.; Yeager, M.; Chanock, S.J.; et al. Risk of non-Hodgkin lymphoma (NHL) in relation to germline variation in DNA repair and related genes. Blood 2006, 108, 3161–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Menashe, I.; Morton, L.M.; Zhang, Y.; Armstrong, B.; Wang, S.S.; Lan, Q.; Hartge, P.; Purdue, M.P.; Cerhan, J.R.; et al. Polymorphisms in DNA repair genes and risk of non-Hodgkin lymphoma in a pooled analysis of three studies. Br. J. Haematol. 2010, 151, 239–244. [Google Scholar] [CrossRef]
- Li, Y.; Bai, O.; Cui, J.; Li, W. Genetic polymorphisms in the DNA repair gene, XRCC1 associate with non-Hodgkin lymphoma susceptibility: A systematic review and meta-analysis. Eur. J. Med. Genet. 2016, 59, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, M.; Travaglino, A.; Varricchio, S.; Russo, D.; Sabattini, E.; Agostinelli, C.; Bertuzzi, C.; Baldo, A.; Pileri, A.; Picardi, M.; et al. Role of chromatin assembly factor-1/p60 and poly [ADP-ribose] polymerase 1 in mycosis fungoides. Virchows Arch. 2021, 478, 961–968. [Google Scholar] [CrossRef]
- Thestrup-Pedersen, K. Cutaneous T-Cell Lymphoma. A hypothesis on disease pathophysiology involving deficiency in DNA repair. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1682–1685. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhao, T.; Cai, J.; Su, Y.; Wang, Z.; Dong, W. Methotrexate induces DNA damage and inhibits homologous recombination repair in choriocarcinoma cells. Onco Targets Ther. 2016, 9, 7115–7122. [Google Scholar] [CrossRef] [Green Version]
- Beksac, M.; Balli, S.; Akcora Yildiz, D. Drug Targeting of Genomic Instability in Multiple Myeloma. Front. Genet. 2020, 11, 228. [Google Scholar] [CrossRef]
- Pawlyn, C.; Loehr, A.; Ashby, C.; Tytarenko, R.; Deshpande, S.; Sun, J.; Fedorchak, K.; Mughal, T.; Davies, F.E.; Walker, B.A.; et al. Loss of heterozygosity as a marker of homologous repair deficiency in multiple myeloma: A role for PARP inhibition? Leukemia 2018, 32, 1561–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathaneni, S.; Stortchevoi, A.; Sommers, J.A.; Brosh, R.M., Jr.; Sharma, S. Human RECQ1 interacts with Ku70/80 and modulates DNA end-joining of double-strand breaks. PLoS ONE 2013, 8, e62481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junior, H.L.R.; de Oliveira, R.T.G.; de Paula Borges, D.; Costa, M.B.; Farias, I.R.; Dos Santos, A.W.A.; Magalhaes, S.M.M.; Pinheiro, R.F. Can synthetic lethality approach be used with DNA repair genes for primary and secondary MDS? Med. Oncol. 2019, 36, 99. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Mohamedali, A.; Eiliazadeh, A.L.; Darling, D.; Mufti, G.J. FLT3 and JAK2 Mutations in Acute Myeloid Leukemia Promote Interchromosomal Homologous Recombination and the Potential for Copy Neutral Loss of Heterozygosity. Cancer Res. 2017, 77, 1697–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhao, J.; Zhang, L.; Tian, S.; Yang, T.; Wang, L.; Zhao, M.; Yang, Q.; Wang, Y.; Yang, X. Evaluation of the Efficacy and Safety of PARP Inhibitors in Advanced-Stage Epithelial Ovarian Cancer. Front. Oncol. 2020, 10, 954. [Google Scholar] [CrossRef]
- Stemmer, A.; Shafran, I.; Stemmer, S.M.; Tsoref, D. Comparison of Poly (ADP-ribose) Polymerase Inhibitors (PARPis) as Maintenance Therapy for Platinum-Sensitive Ovarian Cancer: Systematic Review and Network Meta-Analysis. Cancers 2020, 12, 3026. [Google Scholar] [CrossRef] [PubMed]
- Poggio, F.; Bruzzone, M.; Ceppi, M.; Conte, B.; Martel, S.; Maurer, C.; Tagliamento, M.; Viglietti, G.; Del Mastro, L.; de Azambuja, E.; et al. Single-agent PARP inhibitors for the treatment of patients with BRCA-mutated HER2-negative metastatic breast cancer: A systematic review and meta-analysis. ESMO Open 2018, 3, e000361. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Liu, Y.; Zhang, T.; He, J.; Zhao, H.; An, R.; Xue, Y. Efficacy and safety of PARP inhibitors in the treatment of advanced ovarian cancer: An updated systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Oncol. Hematol. 2021, 157, 103145. [Google Scholar] [CrossRef]
- Xu, Y.; Ding, L.; Tian, Y.; Bi, M.; Han, N.; Wang, L. Comparative Efficacy and Safety of PARP Inhibitors as Maintenance Therapy in Platinum Sensitive Recurrent Ovarian Cancer: A Network Meta-Analysis. Front. Oncol. 2020, 10, 573801. [Google Scholar] [CrossRef]
- Zhou, J.X.; Feng, L.J.; Zhang, X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: A meta-analysis of randomized controlled trials. Drug Des. Devel. Ther. 2017, 11, 3009–3017. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, J. Haematologic toxicities with PARP inhibitors in cancer patients: An up-to-date meta-analysis of 29 randomized controlled trials. J. Clin. Pharm. Ther. 2021, 46, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.D.; Rizzo, A.; Novelli, M.; Tavolari, S.; Palloni, A.; Tober, N.; Abbati, F.; Mollica, V.; De Lorenzo, S.; Turchetti, D.; et al. Specific Toxicity of Maintenance Olaparib Versus Placebo in Advanced Malignancies: A Systematic Review and Meta-analysis. Anticancer Res. 2020, 40, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, A.; Bowering, V.; Ahrari, S.; Oza, A.M.; Lheureux, S. Manage wisely: Poly (ADP-ribose) polymerase inhibitor (PARPi) treatment and adverse events. Int. J. Gynecol. Cancer 2020, 30, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Ruscito, I.; Bellati, F.; Ray-Coquard, I.; Mirza, M.R.; du Bois, A.; Gasparri, M.L.; Costanzi, F.; De Marco, M.P.; Nuti, M.; Caserta, D.; et al. Incorporating Parp-inhibitors in Primary and Recurrent Ovarian Cancer: A Meta-analysis of 12 phase II/III randomized controlled trials. Cancer Treat. Rev. 2020, 87, 102040. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, X.; Zhang, J.; Zhao, Z.; Feng, X.; Liu, L.; Ma, Z. Efficacy and safety of PARP inhibitors in patients with BRCA-mutated advanced breast cancer: A meta-analysis and systematic review. Breast 2021, 60, 26–34. [Google Scholar] [CrossRef]
- Hennes, E.R.; Dow-Hillgartner, E.N.; Bergsbaken, J.J.; Piccolo, J.K. PARP-inhibitor potpourri: A comparative review of class safety, efficacy, and cost. J. Oncol. Pharm. Pract. 2020, 26, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Staropoli, N.; Ciliberto, D.; Del Giudice, T.; Iuliano, E.; Cuce, M.; Grillone, F.; Salvino, A.; Barbieri, V.; Russo, A.; Tassone, P.; et al. The Era of PARP inhibitors in ovarian cancer: “Class Action” or not? A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2018, 131, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Benigno, B.; Dorum, A.; Mahner, S.; Bessette, P.; Barcelo, I.B.; Berton-Rigaud, D.; Ledermann, J.A.; Rimel, B.J.; Herrstedt, J.; et al. Long-term safety in patients with recurrent ovarian cancer treated with niraparib versus placebo: Results from the phase III ENGOT-OV16/NOVA trial. Gynecol. Oncol. 2020, 159, 442–448. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Chamberlain, M.C. High-dose cytarabine salvage therapy for recurrent primary CNS lymphoma. J. Neurooncol. 2016, 126, 545–550. [Google Scholar] [CrossRef]
- Zhu, J.; Tucker, M.; Wang, E.; Grossman, J.S.; Armstrong, A.J.; George, D.J.; Zhang, T. Acute Myeloid Leukemia After Olaparib Treatment in Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, e1137–e1141. [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A., 3rd; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Moore, K.N.; Mirza, M.R.; Matulonis, U.A. The poly (ADP ribose) polymerase inhibitor niraparib: Management of toxicities. Gynecol. Oncol. 2018, 149, 214–220. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; Leary, A.; et al. Patient-Centered Outcomes in ARIEL3, a Phase III, Randomized, Placebo-Controlled Trial of Rucaparib Maintenance Treatment in Patients with Recurrent Ovarian Carcinoma. J. Clin. Oncol. 2020, 38, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.; Sultan, A.; Zaw, M.H.; Thein, K.Z. Secondary hematologic malignancies with poly adenosine diphosphate ribose polymerase inhibitors: Is the buzz real? -Insights from a meta-analysis of phase 3 randomized controlled trials. J. Geriatr. Oncol. 2019, 10, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Thein, K.Z.; Sultan, A.; Zaw, M.H.; Han, M.M.; Yendala, R.; Zin, M.M.; Awasthi, S.; D’Cunha, N.; Hardwicke, F.; Jones, C. Risk of secondary hematological malignancies and hematological toxicities in recurrent ovarian cancer patients treated with poly adenosine diphosphate ribose polymerase (PARP) inhibitors maintenance. Ann. Oncol. 2018, 29, VIII343–VIII344. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of Chemotherapy for Solid Tumors With Development of Therapy-Related Myelodysplastic Syndrome or Acute Myeloid Leukemia in the Modern Era. JAMA Oncol. 2019, 5, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin. Oncol. 2013, 40, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawan, M.S.; Bartelink, I.H.; Aggarwal, R.R.; Leng, J.; Zhang, J.Z.; Pawlowska, N.; Terranova-Barberio, M.; Grabowsky, J.A.; Gewitz, A.; Chien, A.J.; et al. Differential Toxicity in Patients with and without DNA Repair Mutations: Phase I Study of Carboplatin and Talazoparib in Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 6400–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brower, V. Tracking chemotherapy’s effects on secondary cancers. J. Natl. Cancer Inst. 2013, 105, 1421–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]

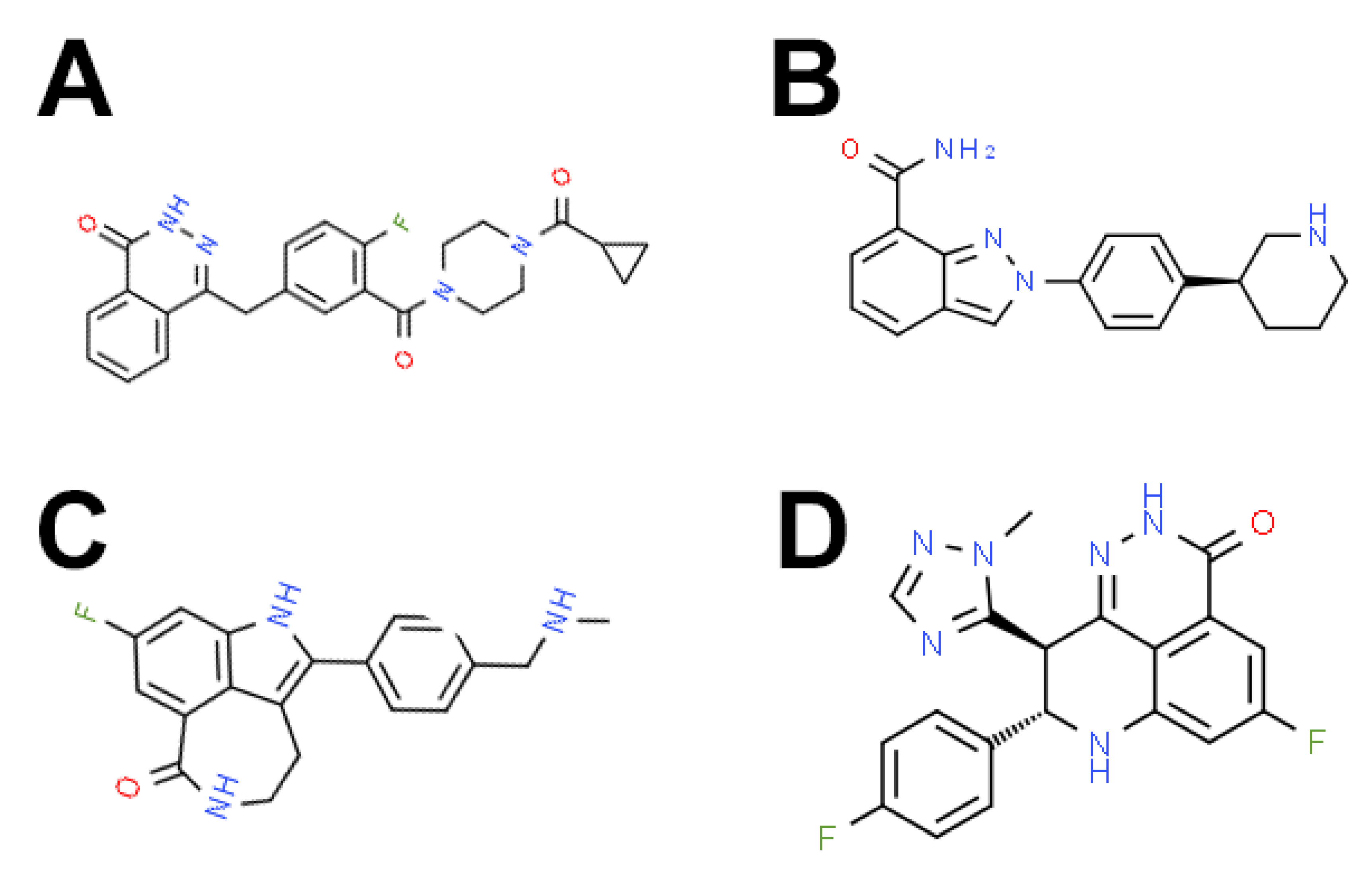

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARP Inhibitor | Other Names | IUPAC/Chemical Name | Target | Haematological Malignancy |
|---|---|---|---|---|
| Olaparib | AZD-2281, KU-59436 | 4-[[3-[4-(cyclopropanecarbonyl)piperazine-1-carbonyl]-4-fluorophenyl]methyl]-2H-phthalazin-1-one | PARP-1/2/3 | ALL, AML, CLL, CML, T-cell lymphoma, MM, MPN, NHL |
| Rucaparib | AG-14699, PF-01367338 | 8-gluoro-2-(4-((methylamino)methyl)phenyl)-4,5-dihydro-1H-azepino[5,4,3-cd]indol-6(3H)-one phosphate | PARP-1/2/3 | ALL, AML, ALL |
| Niraparib | MK4827 | (S)-2-(4-(piperidin-3-yl)phenyl)-2H-indazole-7-carboxamide | PARP-1/2 | AML, NHL |
| Talazoparib | BMN-673, MDV-3800 | ((8S,9R)-5-fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-8,9-dihydro-2H-pyrido[4,3,2-de]phthalazine-3(7H)-one | PARP-1/2 | ALL, AML, CLL, CML, NHL, T-cell lymphoma, MPN |
| Veliparib | ABT-888 | 2-[(2R)-2-methyl-2-pyrrolidinyl]-1H-benzimidazole-7-carboxamide | PARP-1/2 | ALL, AML, CML, MM, MPN, NHL, cutaneous T-cell lymphoma, refractory lymphoma, DLBCL, cHL, FL, tFL |
| CEP-8983 | CK-102 | 11-methoxy-4,5,6,7-tetrahydro-1H-cyclopenta[a]pyrrolo[3,4-c]carboazole-1,3(2H)-dione | PARP-1/2 | CLL |
| PJ34 | - | N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethyl acetamide | PARP-1/2 Phenanthridine PARS inhibitor, tankyrase-1/2 | ALL, AML, CLL, CML, MM |
| 5F02 | - | 1-(2-(cyclododecyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide | PARP-1 (non-NAD-like) | CML |
| AG14361 | - | 1-(4-((dimethylamino)methyl)phenyl)-8,9-dihydro-2,7,9a-triazabenzo[cd]azulen-6(7H)-one | PARP-1 | CML |
| 6-(5H)-phenanthridinone | NSC 11021, NSC 40943, NSC 61083, PHEN | 5H-phenanthridin-6-one | PARP-1 | T-cell lymphoma |
| KU-0058948 | Homopiperazine analogue, 14 | 4-[[4-fluoro-3-[(hexahydro-1H-1,4-diazepin-1-yl)carbonyl]phenyl]methyl]-1(2H)-phthalazinone | PARP | AML, CML |
| NU1025 | NSC 696807 | 8-Hydroxy-2-methyl-4(3H)-quinazolinone | PARP | CML, murine lymphoma |
| AZD-2461 | 1174043-16-3 | 4-(4-fluoro-3-(4-methoxypiperidine-1-carbonyl)benzyl)phthalazine-1(2H)-one | PARP | T-cell lymphoma |
| Cancer | Cell Lines | Drugs | Effect | Mono | Combo | Refs | |
|---|---|---|---|---|---|---|---|
| PARPi | Chemotherapeutic | ||||||
| Acute leukaemias | |||||||
| ALL | MT-4, MT-2, C8166, C91PL, MT-1, ATL-T, ED-40515(-), ALT-25, ATL-43T, KOB, ATL-55T | PJ34 | - | Induced apoptosis and cell cycle arrest | + | N/A | [41] |
| MOLT4 | PJ34 | Vorinostat | PJ34 alone had no effect. Combining PJ34 and vorinostat decreased proliferation | - | ++ | [42] | |
| Jurkat | PJ34 | DAPT | No effect either as monotherapy or in combination with DAPT | - | - | [43] | |
| Patient-derived blasts, Jurkat, MOLT-4, MOLT-3, CCRF-CEM | Olaparib | - | Induced apoptosis | + | N/A | [40] | |
| MOLT3, Jurkat, NALM-6, Reh, KOPN60, KOPN36, YCUB-2, HAL-01, KOCL-58, RS4;11, KOPB-26, KOCL-33 | Olaparib, veliparib | - | Induced apoptosis | + | N/A | [39] | |
| Patient-derived blasts, Jurkat, MOLT-4, HSB2 | Veliparib | TMZ | Veliparib monotherapy inhibited leukaemia cell growth and potentiated the effects of TMZ | + | ++ | [44] | |
| RPMI-8402 | Rucaparib | 5-FU | Sensitive to rucaparib alone, and cell death increased when combined with 5-FU | + | ++ | [45] | |
| NALM-6, COG-LL-317, RS4;11, MOLT4, CCRF-CEM | Talazoparib | TMZ | Combination enhanced cell death | + | ++ | [46] | |
| AML | THP-1, Kasumi-1 | PJ34 | - | PJ34 suppressed proliferation and induced apoptosis | + | N/A | [47] |
| HL60, K562, NB4, U937, Kasumi, OC-1, Raji, KG-1, ME-1, P39, Mutz-3, OCI-AML3, Primary patient samples | KU-0058948, PJ34 | Decitabine, MS275 | Decitabine did not potentiate the effects of PARPi. MS275 enhanced the effects of PARPi in all PARPi sensitive cells | + | Decitabine: - MS275: ++ | [48] | |
| U937, HL60 | PJ34 | Vorinostat | PJ34 alone had no effect. Combining PJ34 and vorinostat decreased proliferation in HL60 cells | - | ++ | [42] | |
| HL60 | PJ34 | Doxorubicin, etoposide, cytarabine, or chlorambucil | PJ34 alone decreased cell survival. Combination did not significantly enhance cytotoxicity | + | ± | [49] | |
| HL-60, U937, NB4, OCI-AML2, OCI-AML3, Patient samples | Olaparib | - | Olaparib induced apoptosis | + | N/A | [50] | |
| HL60 | Olaparib | Gemtuzumab ozogamicin | Olaparib treatment as a monotherapy did not induce apoptosis. Combination with gemtuzumab ozogamicin synergistically enhanced cell death | - | ++ | [51] | |
| KG1a, MV-4-11, PL21, HL60 | Olaparib | Decitabine | Olaparib alone had minimal effect. Combining decitabine and olaparib induced apoptosis | - | ++ | [52] | |
| HL60, MOLM13, THP1, KG1 | Olaparib | Vitamin c | Vitamin C treatment enhanced sensitivity to olaparib | + | ++ | [53] | |
| Patient samples | Olaparib | Avapritinib | Olaparib alone has little effect in patient samples harbouring AML1-ETO and a c-KIT mutation, but induced apoptosis in AML1-ETO AML cells. Combining olaparib and avapritinib increased cell death | AML1-ETO: + AML1-ETO + c-KITMUT: - | ++ | [54] | |
| MOLM13, MV4-11, REH, OCI-AML3 | Olaparib | AZD1775 | Olaparib alone induced cell death. Combining olaparib and AZD1775 significantly enhanced cell death | + | ++ | [55] | |
| THP-1, Kasumi-1, AML patient samples | Olaparib, veliparib | - | AML1-ETO and PML-RARα-driven AML were sensitive to PARPi. MLL-driven AML was not | AML1-ETO, PML-RARα: + MLL: - | N/A | [56] | |
| KG-1, ML-1, Kasumi-1, THP-1, U-937, CMK, NB4, HL60, ML1 | Olaparib, veliparib | - | Not sensitive, with the exception of Kasumi-1 cells | - | N/A | [39] | |
| OCI-AML2 | Rucaparib | 5-FU | Sensitive to rucaparib alone, and combination with 5-FU enhanced cell death | + | ++ | [45] | |
| AML patient blasts; U937, HEL, THP-1, KG-1, HL60 | Veliparib | TMZ | Veliparib monotherapy inhibited leukaemia cell growth and potentiated TMZ | + | ++ | [44] | |
| KBM3/Bu250, MOLM14 | Niraparib | Romidepsin, Panobinostat, decitabine, busulfan, melphalan | Combination synergistically inhibited cell proliferation | + | ++ | [57] | |
| NB4 ATO-sensitive and resistant clones | Olaparib, talazoparib, veliparib, niraparib, rucaparib | Decitabine, azacytidine, ascorbate | All clones were sensitive to olaparib, niraparib and talazoparib, but not to rucaparib and veliparib. Combining PARPi with hypomethylating agents induced synergistic growth inhibitory effects in both ATO-sensitive and resistant clones. Combining ascorbate with niraparib and talazoparib synergistically enhanced effects | Ola: + Nir: + Tal: + Ruc: - Vel: - | ++ | [58] | |
| Kasumi-1, MV-4-11, MOLM13, MOLM14 | Veliparib, talazoparib | Azacytidine | Low doses of azacytidine and PARPi increased cell death | + | ++ | [59] | |
| Patient samples, KG-1, MOLM-13, NB4, OCI-AML2, OCI-AML, P39 | Talazoparib | - | Cells with microsatellite instability were hypersensitive to PARPi | + | N/A | [60] | |
| Kasumi-1 | Talazoparib | TMZ | Combination enhanced cell death | + | ++ | [46] | |
| K563, HL60, Patient samples | Talazoparib | TSA, Entinostat | Talazoparib alone did not induce cell death. Combining with TSA or entinostat significantly increased apoptosis | - | ++ | [61] | |
| MV-4-11, HL60, REH, Patient samples | Olaparib, talazoparib | AC220 | REH cells were highly sensitive to talazoparib. FLT3-ITD cells were more sensitive to olaparib than FLT3-WT cells. Combining with AC220 significantly increased apoptosis in all cells | + | ++ | [62] | |
| Patient samples | Olaparib, talazoparib | - | PARPi-induced apoptosis | + | N/A | [63] | |
| Patient samples, K562, Kasumi-1 | Olaparib, talazoparib | SB431542 | Cells were sensitive to olaparib and talazoparib. When co-cultured with bone marrow stromal cells, AML cell sensitivity to PARPi decreased. This was restored by combined treatment with SB435142 | + Co-culture: - | ++ | [64] | |
| Patient samples (IDH1/2MUT and IDH1/2WT) | Olaparib, talazoparib | Daunorubicin, irradiation, AGI-5198, AGI-6780 | IDH1/2 mutant cells were sensitive to PARPi, and this was enhanced via combination with daunorubicin or irradiation. IDH1/2 inhibitors antagonised this effect | + | DNR: ++ Irrad: ++ IDH inhibitors: -- | [65] | |
| Chronic leukaemia | |||||||
| CML | K562 | PJ34 | Vorinostat | PJ34 had no effect. Combining PJ34 and vorinostat decreased proliferation | - | ++ | [42] |
| K562 | KU-0058948, PJ34 | Decitabine, MS275 | PARPi-induced cell cycle arrest and apoptosis. Addition of decitabine failed to increase cytotoxicity of PARPi. Combination with MS275 potentiated the cytotoxic effect of PARPi | + | Dec: - MS275: ++ | [48] | |
| K562 | AG14361 | Camptothecin | AG14361 enhanced the growth inhibitory and cytotoxic effects of camptothecin | N/A | ++ | [66] | |
| K562, K562 imatinib resistant, Mo7e-P210 IMR2 | NU1025 | L67 | Cells were sensitive to NU1025, and combining with L67 significantly increased cell death, even in imatinib resistant cell lines | + | ++ | [67] | |
| Patient samples | 5F02, olaparib, talazoparib | - | PARPi-induced apoptosis as single agents. Combining 5F02 and olaparib induced apoptosis in quiescent CML cells | + | N/A | [68] | |
| K562 | Olaparib | Decitabine | Olaparib alone had minimal effect. Combining decitabine and olaparib induced apoptosis | - | ++ | [52] | |
| BV173, K562, AR230, MEG-01 | Olaparib, veliparib | - | Induced apoptosis in all cell lines except K562 | + | N/A | [39] | |
| K562, MEG01 | Olaparib | Cisplatin | Olaparib blocked cisplatin-induced cell death | + | -- | [69] | |
| Patient samples grown on a HS-5 stromal cell monolayer | Talazoparib | Imatinib | Talazoparib inhibited the clonogenic potential of imatinib-refractory CML cells | + | ++ | [70] | |
| Paediatric patient samples | Talazoparib | Chloroquine | Talazoparib monotherapy induced cytotoxicity. Combination synergistically enhanced cell death | + | ++ | [71] | |
| Lymphomas | |||||||
| CLL | CLL, 697 | PJ34 | DAPT | No effect either as monotherapy or in combination with DAPT | - | - | [43] |
| Patient samples | CEP-8983 | Bendamustine | CEP-8983 induced cytotoxicity as a single agent. Synergistically enhanced cell death when combined with bendamustine | + | ++ | [72] | |
| Patient samples, PGA (parental and ATM knockdown) | Olaparib | 4HC, fludarabine, valproic acid, bendamustine and irradiation | ATM-deficient cells were sensitive to olaparib. Olaparib sensitised cells to treatment with 4HC, fludarabine, valproic acid, bendamustine and irradiation | + | ++ | [73] | |
| Patient samples | Talazoparib | - | Induced cell death | + | N/A | [74] | |
| NHL | Granta-519, Jeko1, JVM2, Z138C | AG14361 | Topotecan | AG14361 potentiated topotecan cytotoxicity independently of TP53 or either ATM or BRCA2 knockdown/inhibition | N/A | ++ | [75] |
| Granta-519, HBL-2, JVM-2, MAVER-1, Z138, C35ABR, L3 | Olaparib, PJ34 | - | Induced cell death in ATM-deficient cells | + | N/A | [76] | |
| C35ABR, UPN1, UPN2, Granta-519, HBL-2, JVM-2, Z138, L3 | Olaparib | - | Induced cell death in ATM-deficient cells | + | N/A | [77] | |
| Raji, Daudi | Olaparib, veliparib | - | Induced apoptosis | + | N/A | [39] | |
| Mutu, Raji, DG75, and patient samples | Olaparib, talazoparib | - | Decreased cell survival | + | N/A | [78] | |
| Raji | Olaparib, veliparib | External source caesium-based radiation and 131I-tositumomab | PARPi sensitised cells to irradiation | + | ++ | [79] | |
| OCI-LY1, OCI-LY8, SUDHL-6, G452, VAL, DOHH2, U2932, OCI-LY19, HCC1187 | Olaparib | Doxorubicin | Olaparib alone decreased proliferation and colony formation in LMO2 expressing cells. Combination with doxorubicin synergistically enhanced these effects. | + | ++ | [80] | |
| Granta-519, JVM-2 | Olaparib | 4HC, fludarabine, valproic acid, bendamustine and irradiation | ATM-deficient cells were more sensitive to olaparib that ATM competent cells. Olaparib sensitised cells to treatment with 4HC, fludarabine, valproic acid, bendamustine and irradiation | + | ++ | [73] | |
| Granta-519, Z-138 | Olaparib | Ibrutinib | Olaparib exhibited cytotoxicity as a single agent. Combining olaparib and ibrutinib synergistically enhanced cell death | + | ++ | [81] | |
| Toledo | Niraparib | Romidepsin, Panobinostat, decitabine, busulfan, melphalan | Combination synergistically inhibited cell proliferation | + | ++ | [57] | |
| Karpas-299, RAMOS-RA1 | Talazoparib | TMZ | Enhanced cell death with combination | + | ++ | [46] | |
| T-cell lymphoma | Sezary Syndrome Patient samples | AZD-2461 | - | Induced cell death | + | N/A | [82] |
| RDM4 | 6(5H)-phenanthridinone | 60Co panoramic γ-radiation | PARPi sensitised cells to irradiation | + | ++ | [83] | |
| J45.01, Toledo, T-cell lymphoma Patient samples | Olaparib | Gemcitabine, busulfan, melphalan | Olaparib enhanced the cytotoxicity of combined gemcitabine, busulfan, and melphalan | + | ++ | [84] | |
| MBL2, HUT78, EL4, | Talazoparib | Bexarotene, vorinostat, romidepsin, methotrexate, pralatrexate, bortezomib | Talazoparib arrested the cell cycle at G2/M. Combining talazoparib with romidepsin or bexarotene synergistically enhanced cell death. Combining talazoparib with vorinostat, methotrexate, pralatrexate or bortezomib exhibited antagonistic effects. | + | Rom: ++ Bex: ++ Vor: -- Meth: -- Pra: -- Bort: -- | [85] | |
| Multiple Myeloma | |||||||
| XG7, XG19 | PJ34 | - | PJ34 treatment slightly decreased cell viability | + | N/A | [86] | |
| RPMI8226/R, RPMI8226 | PJ34 | Melphalan | PJ34 enhanced the cytotoxicity of melphalan in RPMI8226/R cells but not RPMI8226 cells | + | ++ | [87] | |
| RPMI-8226, MM1.S | PJ34 | Dexamethasone | PJ34 enhanced the cytotoxicity of dexamethasone | + | ++ | [88] | |
| CAPAN1, H929, OPM2, U266m R8226, INA6, KMS26, M12BM, KMS11, bortezomib resistant AMO1 cells | Olaparib | - | Majority of cell lines (except U266, KMS11, OPM2) were sensitive to olaparib | + | N/A | [89] | |
| NCI-H929, RPMI-8226, MM.1S, normal human peripheral CD19+ B cells | Veliparib | Dinaciclib | Veliparib alone did not induce cell death. Combining with dinaciclib induced synthetic lethality in MM cells, but not normal peripheral B cells | - | ++ | [90] | |
| NCI-H929, RPMI-8226, KMS11, MM1S, OPM2 | Veliparib | Bortezomib | Veliparib alone had limited effect, but combining with bortezomib significantly increased cell death | - | ++ | [91] | |
| Myeloproliferative Neoplasms | |||||||
| BCR/ABL+ CML, ET, PV, primary and secondary MF and mixed MDS/MPN patient samples | Veliparib, olaparib | - | MPN patient samples with impaired RAD51 foci were particularly sensitive to treatment with PARPi | + | N/A | [92] | |
| MF patient samples | Veliparib | Busulfan | MF patient samples were sensitive to PARPi treatment alone. Combining with busulfan significantly enhanced these effects | + | ++ | [93] | |
| JAK2V617F, CALR (del52), MPL (W515L) patient samples | Olaparib, talazoparib | Ruxolitinib, hydroxyurea | MPN samples were sensitive to PARPi treatment. Combining with ruxolitinib and hydroxyurea significantly enhanced sensitivity of MPN cells to PARPi | + | ++ | [94] | |
| Patient samples | Olaparib | Decitabine | Olaparib alone induced cytotoxicity and was enhanced by combining with decitabine | + | ++ | [95] | |
| Cancer | Model | Treatment Regimen | Effect | Mono | Combo | Refs | |
|---|---|---|---|---|---|---|---|
| PARPi | Chemotherapeutic | ||||||
| Acute Leukaemias | |||||||
| ALL | NSG mice, patient-derived T-ALL samples pre-treated with olaparib i.v. | Pre-treatment with 5µM olaparib for 48 h prior to transplantation | - | Decreased engraftment | + | N/A | [40] |
| Female NSG mice, HAL-01 or YCUB-2 cells i.v. | Olaparib (100 mg/kg) orally 5 times per week | TMZ (25 mg/kg) orally 5 times per week | Olaparib and TMZ alone no effect. Combination significantly increased survival | - | ++ | [39] | |
| (1) Female and male C57BL6/Ly5.1 mice transplanted with spleen cells from ALL ENU treated mice (2) Female and male NSG mice, patient-derived T-ALL sample i.v. | (1) Rucaparib (1 mg/kg) i.p. for 5 days (2) Rucaparib (1.3 mg/kg) i.p. for 5 days | (1) 5-FU (150 mg/kg) i.p. on day 2 (2) 5-FU (75 mg/kg) i.p. on day 2 | Rucaparib alone had no effect in either model. Combination with 5-FU increased survival in both models | - | ++ | [45] | |
| Female NOD.CB17-Prkdcscid/J mice, 8 patient-derived ALL samples i.v. | Talazoparib (0.25 mg/kg) twice daily | TMZ (12 mg/kg) daily × 5 | No objective response, and excessive toxicity for 7/8 xenografts | - | - | [46] | |
| NSG mice, patient-derived ALL cells i.v. | Talazoparib (0.33 mg/kg) orally for 7 days | Imatinib (100 mg/kg) orally twice daily | Talazoparib alone increased survival. Combination with imatinib further increased survival | + | ++ | [63] | |
| AML | Male C57Bl/6J mice, murine C1498 cells i.v. | PJ34 (10 mg/kg) i.p. daily for four weeks | - | PJ34 increased survival and delayed tumour progression | + | N/A | [47] |
| Female and male NSG mice, (1) Kasumi cells i.f., (2) THP-1 i.v., (3) patient-derived MLL-AML samples i.f. | (1, 2) Olaparib (25 mg/kg) i.p. daily for 2–4 weeks (3) Olaparib (25 mg/kg) i.p. every other day for 4 weeks | (1) - (2) - (3) 0.4% lithium carbonate-containing diet | (1) Olaparib increased survival in Kasumi model. (2, 3) Olaparib did not increase survival in THP-1 or MLL-driven AML PDX models. (3) Combination increased survival | (1) + (2, 3) - | ++ | [56] | |
| Female C57BL/6J mice, AML cells i.v. | Olaparib (50 mg/kg) orally for 5 days | AZD1775 (80 mg/kg) orally for 5 days | Olaparib alone slightly extended survival. Combination significantly improved survival and reduced tumour burden | + | ++ | [55] | |
| Female and male NSG mice, patient-derived M4-AML sample i.v. | Rucaparib (1.3 mg/kg) i.p. daily for 5 days | 5-FU (150 mg/kg) i.p. on day 2 | Rucaparib alone did not increase survival. Combination with 5-FU increased survival | - | ++ | [45] | |
| Female NSG mice, MOLM-14 or MV-4-11 i.v. | Talazoparib (0.1 mg/kg) orally 5 days per week | Azacytidine (0.5 mg/kg) s.c. 5 days per week | Combination decreased tumour burden and increased survival | - | ++ | [59] | |
| NSG mice, patient-derived AML cells i.v. | Talazoparib (0.33 mg/kg) orally for 7 days | Doxorubicin (1.5 mg/kg) i.v. on days 1–3 and cytarabine (50 mg/kg) i.v. on days 1–5 | Talazoparib alone did not increase survival. Combination with doxorubicin and cytarabine increased survival | - | ++ | [63] | |
| NRGS mice, patient-derived AML-FLT3ITD sample i.v. | Talazoparib (0.33 mg/kg) daily for 7 days | AC220 (10 mg/kg) for days | Talazoparib alone had no effect. Combining talazoparib and AC220 significantly increased survival | - | ++ | [62] | |
| Female NOD.CB17-Prkdcscid/J mice, patient-derived FLT3-ITD samples i.v. or Tet2−/− AML-like murine leukaemias i.v. | Talazoparib (0.165 mg/kg) i.v. for 7 days | Imatinib (100 mg/kg) daily for 7 days or Quizartinib (1 mg/kg) daily for 7 days | Combined PARPi and imatinib or Quizartinib increased survival | N/A | ++ | [64]. | |
| NSGS mice, U937 wild-type and STAG2-knockout | Talazoparib (0.25 mg/kg) oral daily | - | Decreased leukaemic burden in STAG2 knockout cells | + | N/A | [96] | |
| Female C57BL/6 mice, RFP-GFP-double positive leukaemic spleen cells from mice that underwent secondary/ tertiary transplantation that developed acute erythroid leukaemia | Talazoparib (0.1 mg/kg) oral days 1–5 and days 14–19 | Decitabine (0.5 mg/kg) i.v. daily days 1–5 | Combination with decitabine significantly decreased spleen size, but not survival | + | ++ | [97] | |
| Chronic Leukaemias | |||||||
| CML | NSG mice, patient-derived CML-CP or CML-AP samples i.v. | Talazoparib (0.33 mg/kg) orally for 14 days | Imatinib (100 mg/kg) orally twice daily | Talazoparib alone increased survival. Combination with imatinib significantly increased this effect | + | ++ | [63] |
| NOD.Rag1−/−;γcnull mice, patient-derived CML-CP samples i.v. | 5F02 (2.5 mg/kg) i.p. and/or Talazoparib (0.33 mg/kg) i.v. | Imatinib (100 mg/kg) orally twice daily 7 days and | Combined administration of imatinib and PARPi reduced tumour burden | N/A | ++ | [68] | |
| Total body irradiated (600 cGy) mice, patient-derived CML-CP samples pre-treated with drugs i.v. | Pre-treatment with talazoparib (100 nM) | Pre-treatment with imatinib (1 µM) | Pre-treatment with the combination prevented engraftment | + | ++ | [70] | |
| Male BALB/c nude mice, patient-derived CML sample s.c. | Talazoparib (50 mg/kg) orally daily | Chloroquine (50 mg/kg) i.p. daily | Talazoparib decreased tumour burden. Combination with chloroquine synergistically decreased tumour volume | + | ++ | [71] | |
| Lymphomas | |||||||
| CLL | Conditional deletion of ATM in B-cells in Eµ: TCL1 mice on a mixed C57BL/6J-C57BL/6N background | Olaparib (50 mg/kg) i.p. 5 days per week | - | Increased survival and improved spleen volume | + | N/A | [98] |
| Murine Lymphoma | Male C57BL/6 × DBA/2 mice, L5178Y cells intracranially | NU1025 (1 mg/mouse) intracranially | TMZ (100 mg/kg or 200 mg/kg) i.p. | NU1025 alone had no effect on survival. Combining with TMZ significantly increased survival | - | ++ | [99] |
| NHL | NOD/SCID mice, Granta-519 i.v. | Olaparib (50 mg/kg) i.p. for 14 days | - | Olaparib decreased tumour engraftment and increased survival | + | N/A | [73] |
| Female RAG2−/− mice, Granta-519 or Z138 s.c. | Olaparib (25 mg/kg or 50 mg/kg) i.p. for 28 days | - | Delayed tumour growth and increased survival | + | N/A | [76] | |
| Female RAG2−/− mice, Granta-519 or UPN2 s.c. | Olaparib (50 mg/kg) i.p. for 28 days | - | Delayed tumour growth and increased survival | + | N/A | [77] | |
| (1) NOD-SCID mice, OCI-LY19, OCI-LY1, or OCI-LY8 cells s.c., (2) NSG mice, patient-derived samples s.c. | Olaparib (50 mg/kg) i.p. daily | R-CHOP: Rituximab (20 mg/kg) + cyclophosphamide (40 mg/kg) + Doxorubicin (3.3 mg/kg) + vincristine (0.5 mg/kg) i.v. on day 1, prednisone (0.2 mg/kg) orally for 5 days | Olaparib alone increased survival in all models. When combined with R-CHOP in OCI-LY8 xenografts delayed tumour growth and significantly improved survival | + | ++ | [80] | |
| Female NSG mice, patient-derived Burkitt lymphoma samples i.v. | Talazoparib (0.33 mg/kg) orally for 7 days | Cytarabine (50 mg/kg) i.v. on days 1–5 | Talazoparib monotherapy decreased tumour burden and increased survival. Combining talazoparib and cytarabine synergistically enhanced effects | + | ++ | [78] | |
| Cutaneous lymphoma | C57Bl/6 mice, MBL cells dermal injection | (1) Talazoparib (dose not reported) i.p. for 7 days (2) Talazoparib (dose not reported) orally for 7 days | (1) - (2) Romidepsin (dose not reported) i.p. for 7 days | Talazoparib treatment significantly reduced tumour volume. Combination with romidepsin significantly increased this effect | + | ++ | [85] |
| Multiple Myeloma | |||||||
| Male CB-17 SCID mice, H929 or bortezomib resistant AMO1 cells s.c. | Olaparib (100 mg/kg) orally daily | - | Reduced tumour growth in both xenograft models | + | N/A | [89] | |
| CB-17 SCID mice, MM.1S cells s.c. | Veliparib (50 mg/kg) orally twice daily | Bortezomib (0.4 mg/kg) s.c. twice per week | Combination decreased tumour burden and increased survival | - | ++ | [91] | |
| Male CB-17 SCID mice, MM.1S cells s.c. | Veliparib (50 mg/kg) orally twice daily, 5 days per week | Dinaciclib (35 mg/kg) i.p. twice per week | Veliparib alone had no effect on tumour volume or survival. Combining with dinaciclib delayed tumour growth and improved survival | - | ++ | [90] | |
| Myeloproliferative Neoplasms | |||||||
| Murine MPN-like disease | C57BL/6 mice injected with GFP+JAK2V617F and WT bone marrow cells | Talazoparib (0.33 mg/kg) i.v. daily | Hydroxyurea (30 mg/kg) i.p. twice daily, and/or ruxolitinib (30 mg/kg) orally twice daily | Talazoparib decreased tumour burden. Combining with hydroxyurea and/or ruxolitinib significantly increased these effects | + | ++ | [94] |
| ET | NSG mice, SET2 cells i.v. | Veliparib (3 mg/kg) i.p. for 5 days | Busulfan (25 mg/kg) i.p. weekly | Veliparib alone had no effect on survival, but combining with busulfan significantly increased survival | - | ++ | [93] |
| MDS | Sequential bone marrow transplant to generate Tet2/STAG2 mutant mice | Talazoparib (0.25 mg/kg) orally daily | - | Talazoparib selectively depleted cohesin-mutant cells | + | N/A | [96] |
| Cancer Types | Clinical Trials Identifier | Phase | Treatment Regimen | Effects | Refs |
|---|---|---|---|---|---|
| Advanced solid tumours (n = 4), NHL (n = 3), cutaneous T-cell lymphoma (n = 2) | - | Phase 0 | Veliparib (10, 25 or 50 mg) single oral dose | Veliparib treatment decreased PAR and the ratio of PAR to PARP-1 in tumour cells | [116] |
| Advanced solid tumours (n = 8), low grade lymphoma (n = 3), cutaneous T-cell lymphoma (n = 3) | NCT00387608 | Phase 0 | Veliparib (10, 25, 50, 100, or 150 mg) single oral dose | Good oral bioavailability and was well tolerated. Significant inhibition of PAR levels in tumour cells at the 25 and 50 mg doses | [117] |
| Colorectal cancer (n = 5), ovarian (n = 5), melanoma (n = 2), pancreas (n = 1), endometrial cancer (n = 1), Hurthle cell thyroid (n = 1), other (n = 8; pleural mesothelioma, hepatocellular, NHL, external ear adenocarcinoma, bile duct adenocarcinoma, small-cell lung cancer, oesophageal adenocarcinoma, chondrosarcoma) | NCT00553189 | Phase I | Cohort 1: 10 mg po BID veliparib days 1–7 + 1.2 mg/m2/d i.v. topotecan days—8, 2–5 (cycle 1) and days 1–5 (cycle 2 onwards (n = 6); Cohort 2: 10 mg po BID veliparib days 1–7 + 0.9 mg/m2/d i.v. topotecan days—8, 2–5 (cycle 1) and days 1–5 (cycle 2 onwards (n = 3); Cohort 3: -2: 10 mg po BID veliparib days 2–5 (cycle 1) and days 1–5 (cycle 2 onwards) + 0.75 mg/m2/d i.v. topotecan days 1–5 (n = 3); Cohort 4: −3: 10 mg po BID veliparib days 2–5 (cycle 1) and days 1–5 (cycle 2 onwards) + 0.6 mg/m2/d i.v. topotecan days 1–5 (n = 4 + 3); Cohort 5: 10 mg po BID veliparib day 1 + 0.75 mg/m2/d i.v. topotecan days 1–5 (n = 5) | Most common DLTs: grade 4 neutropaenia and thrombocytopaenia, grade 4 neutropaenia lasting >5 days, febrile neutropaenia, grade 3 or 4 myelosuppression. Four of 6 patients (66.7%) on dose level 1 had stable disease after 2 cycles (taken off study due to toxicity) | [118] |
| Advanced solid tumours (n = 33) and refractory lymphoma (n = 2) | NCT00810966 | Phase I | Cohort 1: 20 mg veliparib QD × 7 days + 50 mg cyclophosphamide QD × 21 days (n = 3); Cohort 2: 30 mg veliparib QD × 7 days + 50 mg cyclophosphamide QD × 21 days (n = 3); Cohort 3: 30 mg veliparib QD × 14 days + 50 mg cyclophosphamide QD × 21 days (n = 3); Cohort 4: 40 mg veliparib QD × 21 days + 50 mg cyclophosphamide QD × 21 days (n = 3); Cohort 5: 40 mg veliparib QD × 21 days + 100 mg cyclophosphamide QD × 21 days (n = 3); Cohort 6: 50 mg veliparib QD × 21 days + 50 mg cyclophosphamide QD × 21 days (n = 3); Cohort 7: 60 mg veliparib QD × 21 days + 50 mg cyclophosphamide QD × 21 days (n = 14); Cohort 8: 80 mg veliparib QD × 21 days + 50 mg cyclophosphamide QD × 21 days (n = 3) | Generally well tolerated. Grade 2 myelosuppression was the most common toxicity. Twelve out of 35 (34%) patients exhibited grade 3 or 4 lymphopaenia. Seven out of 35 (20%) of patients experienced a partial response, and 6/35 (17%) exhibited prolonged stable disease, including 1 patient with lymphoma | [119] |
| De novo or secondary AML (n = 48) | NCT01139970 | Phase I | Veliparib 20–200 mg PO day 1 and BID days 5–12 (cycle 1) or days 1–8 (cycle 2 onwards) + 150–200 mg/m2/d PO days 3–8 (cycle 1) or days 1–5 (cycle 2 onwards) every 28–56 days | No DLT observed for 20–150 mg veliparib. A DLT of grade 3 oropharyngeal mucositis/esophagitis was observed in 2/4 (50%) of patients treated with 200 mg veliparib. The most common serious adverse events were infections (40%), febrile neutropaenia (25%), and oropharyngeal mucositis/esophagitis (4%). Complete responses were attained in 8/48 (16.6%) patients, with 7/8 achieving complete remission after a single cycle. An additional 8/48 (16.6%) patients exhibited disease stabilisation | [115] |
| Relapsed refractory AML, newly diagnosed aggressive MPN, aggressive CMMoL (n = 99) | NCT03289910 | Phase I | Veliparib PO BID with dose escalation (10–100 mg × 8 days; 80 mg × 14 or 21 days) + 1–1.3 mg/m2/d topotecan continuous i.v. days 3–7 + 120–150 mg/m2/d carboplatin continuous i.v. days 3–7 | Response rate in AML with no history of MPN or CMMoL was 25% (19/77). Response rate in aggressive MPN, CMMoL or related AML was 64% (14/22). Mucositis was dose limiting and correlated with high veliparib concentrations | [120] |
| Relapsed/refractory lymphoma (n = 23; cHL, DLBCL, FL, tFL), MM (n = 1), and advanced solid malignancies (n = 25) | NCT01326702 | Phase I/II | Dose-escalation cohorts (n = 34): 200–400 mg PO BID veliparib days 1–7 of 28-day cycle + 70 and 90 m2/d bendamustine i.v. days 1 and 2; Cohort expansion (n = 7) in B-cell lymphomas: 300 mg PO BID veliparib + 90 mg/m2 bendamustine and 375 mg/m2 rituximab day 1 | DLTs: anaemia, nausea, hypertensions, and hyperhidrosis. Most common grade 3–4 toxicity was lymphopaenia (88%), anaemia (20%), neutropaenia (12%), thrombocytopaenia (10%), and leucopoenia (10%). Five of seven (71%) lymphoma patients on veliparib + bendamustine and 6/7 (86%) on veliparib + bendamustine + rituximab achieved objective response. Patient with MM achieved a partial response | [121] |
| Relapsed CLL (n = 9), MCL (n = 4) and T-PLL (n = 2) | ISRCTN34386131 | Phase I | Cohort 1: 200 mg capsule olaparib PO BID (n = 6); Cohort 2: 400 mg olaparib capsule PO BID (n = 3); Cohort 3: 100 mg olaparib tablet PO BID (n = 6) | Myelosuppression was the most common haematological grade 3–4 toxicity (n = 8). Overall, both formulations of olaparib were well tolerated, with the most common AEs being anaemia (66%), thrombocytopaenia (53%), fatigue (53%), nausea (33%) and neutropaenia (20%). All 3 patients who received the higher dose of 400 mg BID capsules developed DLTs possibly attributable to olaparib. The median OS for patients treated with capsules (106 days) was not dissimilar to that for patients treated with tablets (129 days) | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skelding, K.A.; Lincz, L.F. PARP Inhibitors and Haematological Malignancies—Friend or Foe? Cancers 2021, 13, 5328. https://doi.org/10.3390/cancers13215328
Skelding KA, Lincz LF. PARP Inhibitors and Haematological Malignancies—Friend or Foe? Cancers. 2021; 13(21):5328. https://doi.org/10.3390/cancers13215328
Chicago/Turabian StyleSkelding, Kathryn A., and Lisa F. Lincz. 2021. "PARP Inhibitors and Haematological Malignancies—Friend or Foe?" Cancers 13, no. 21: 5328. https://doi.org/10.3390/cancers13215328