
Somatic Hypomethylation of Pericentromeric SST1 Repeats and Tetraploidization in Human Colorectal Cancer Cells

 , , ,
, , ,  ,
,  and
and
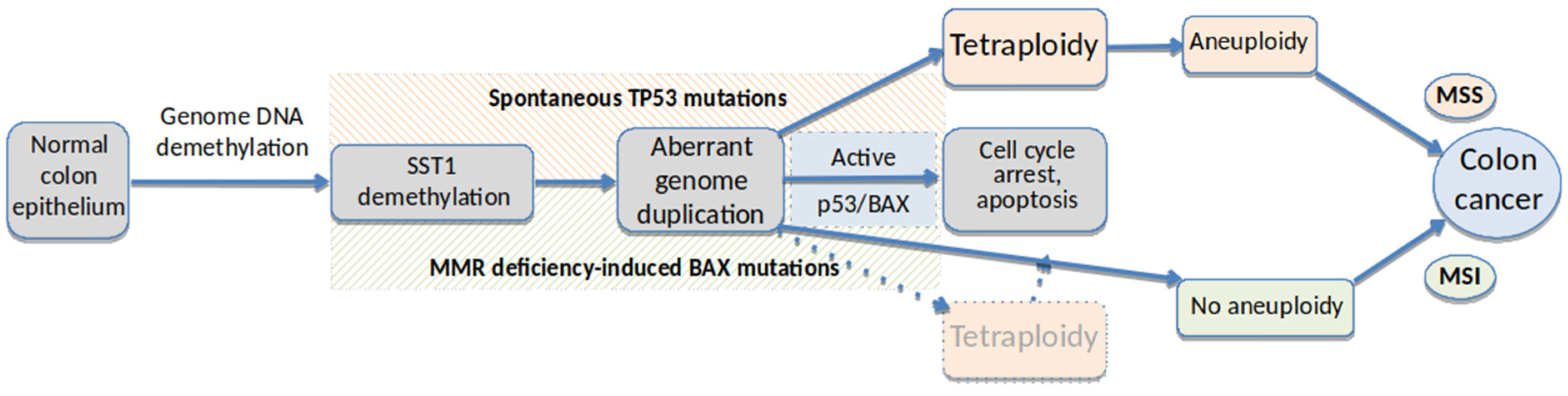
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. DNA Methylation Alterations in Cancer
1.2. Hypomethylation of SST1 Pericentromeric Repeats in Cancer
1.3. The Role of Aneuploidy in Cancer
1.4. The Importance of Tetraploidy in Cancer
1.5. TP53 and Aneuploidy in Cancer
2. Materials and Methods
2.1. Cell Lines and Tumor Samples
2.2. Cell Culture Conditions
2.3. Isolation of Single Cell-Derived LS174T Cell Clones
2.4. Nuclei Size Measurement
2.5. Karyotyping
2.6. DNA and RNA Extraction
2.7. LINE-1 MS-QPCR
2.8. SST1 MS-QPCR
2.9. Bisulfite Sequencing
2.10. Illumina HM450K Methylation Arrays
2.11. Gene Expression Arrays and Gene Set Enrichment Analysis
2.12. Statistical Analyses
3. Results
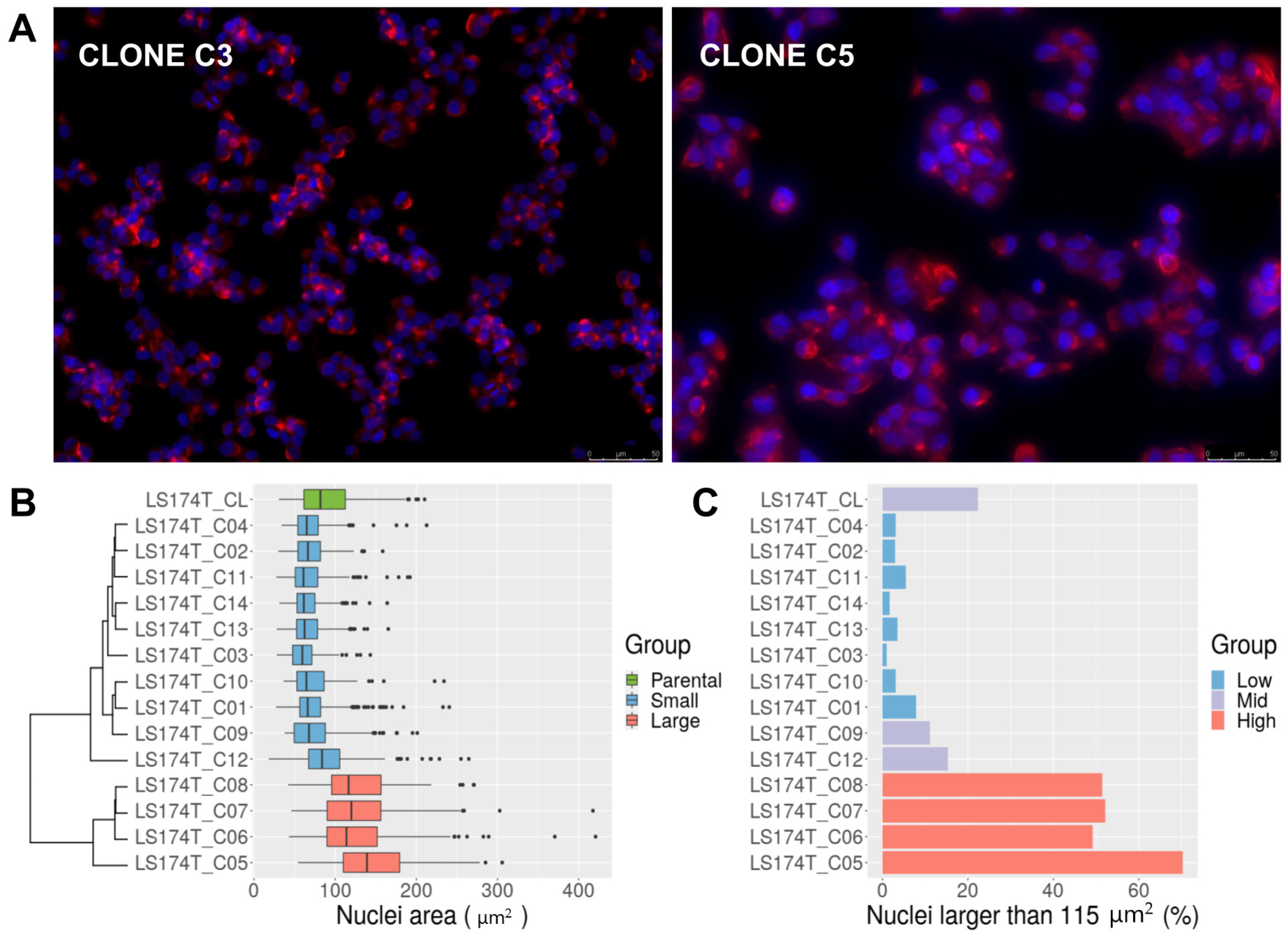
3.1. Isolation of LS174T Single Cell-Derived Clones
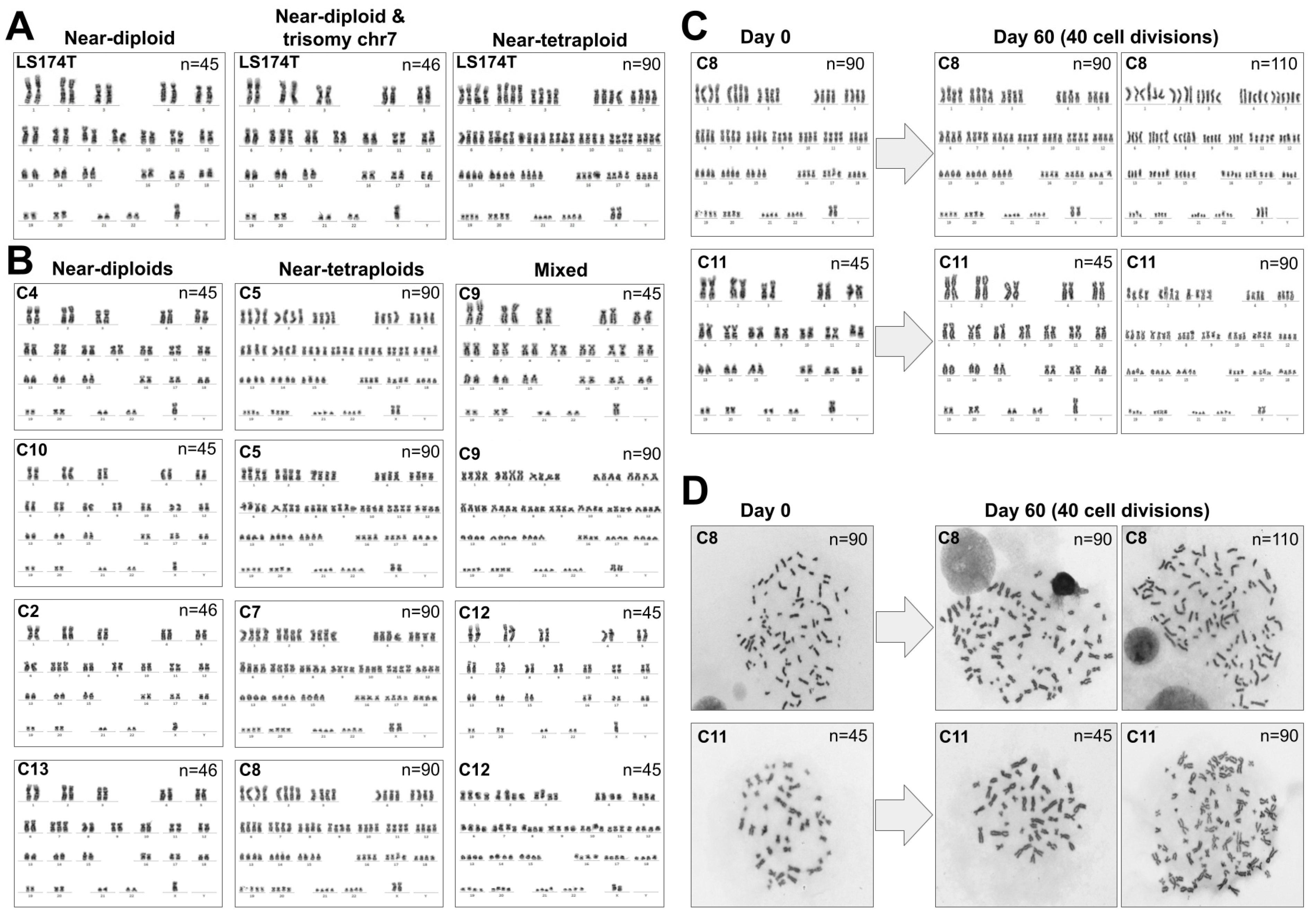
3.2. LS174T Is a Mixture of Normal-Sized and Large-Nucleus Cells with Different Ploidy
3.3. Analysis of Other Cancer Cell Lines
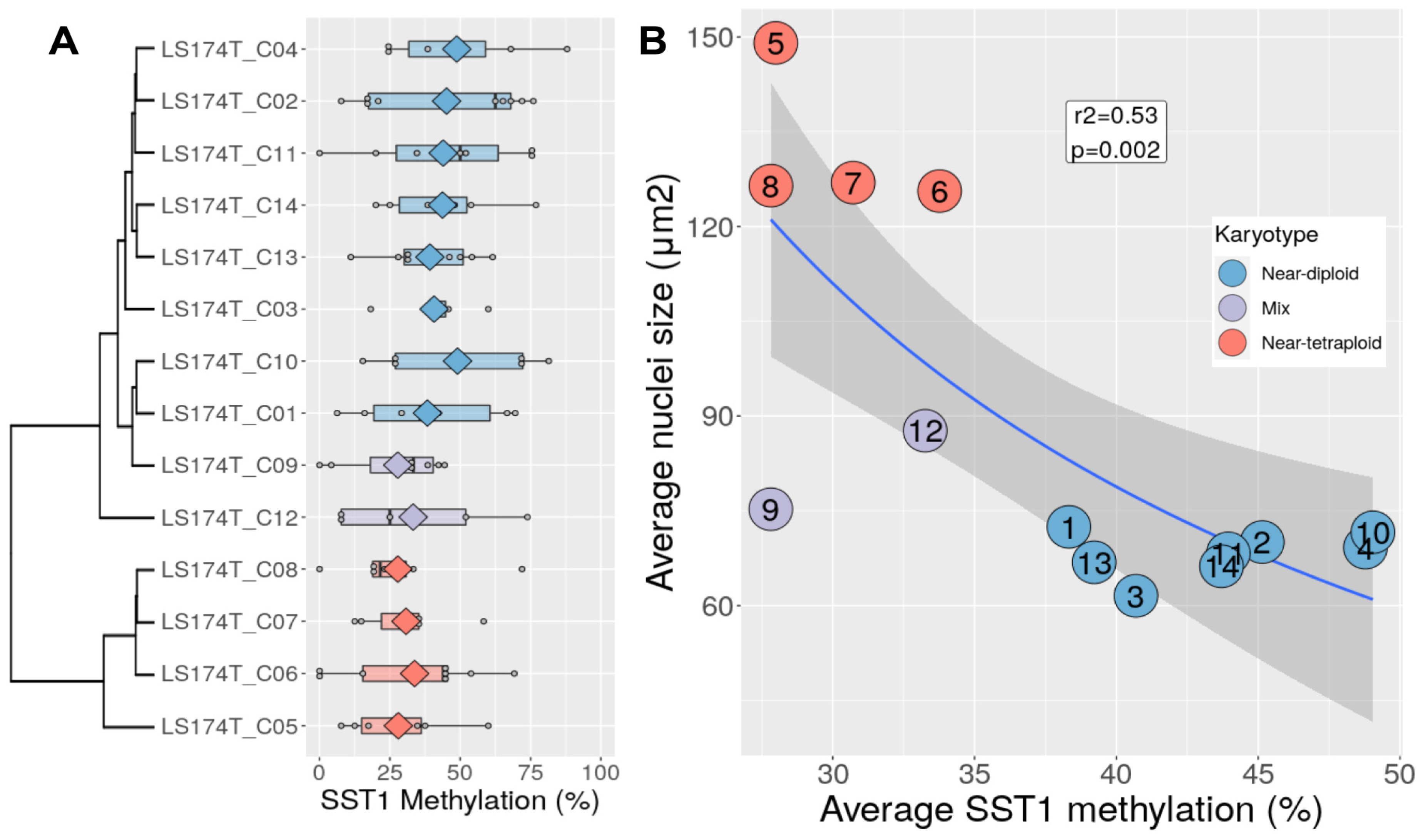
3.4. Chromosomal Content Correlates with SST1 Methylation Level in LS174T Cells
3.5. Transcriptional Profile Differences between Near-Diploid and Near-Tetraploid LS174T Cells
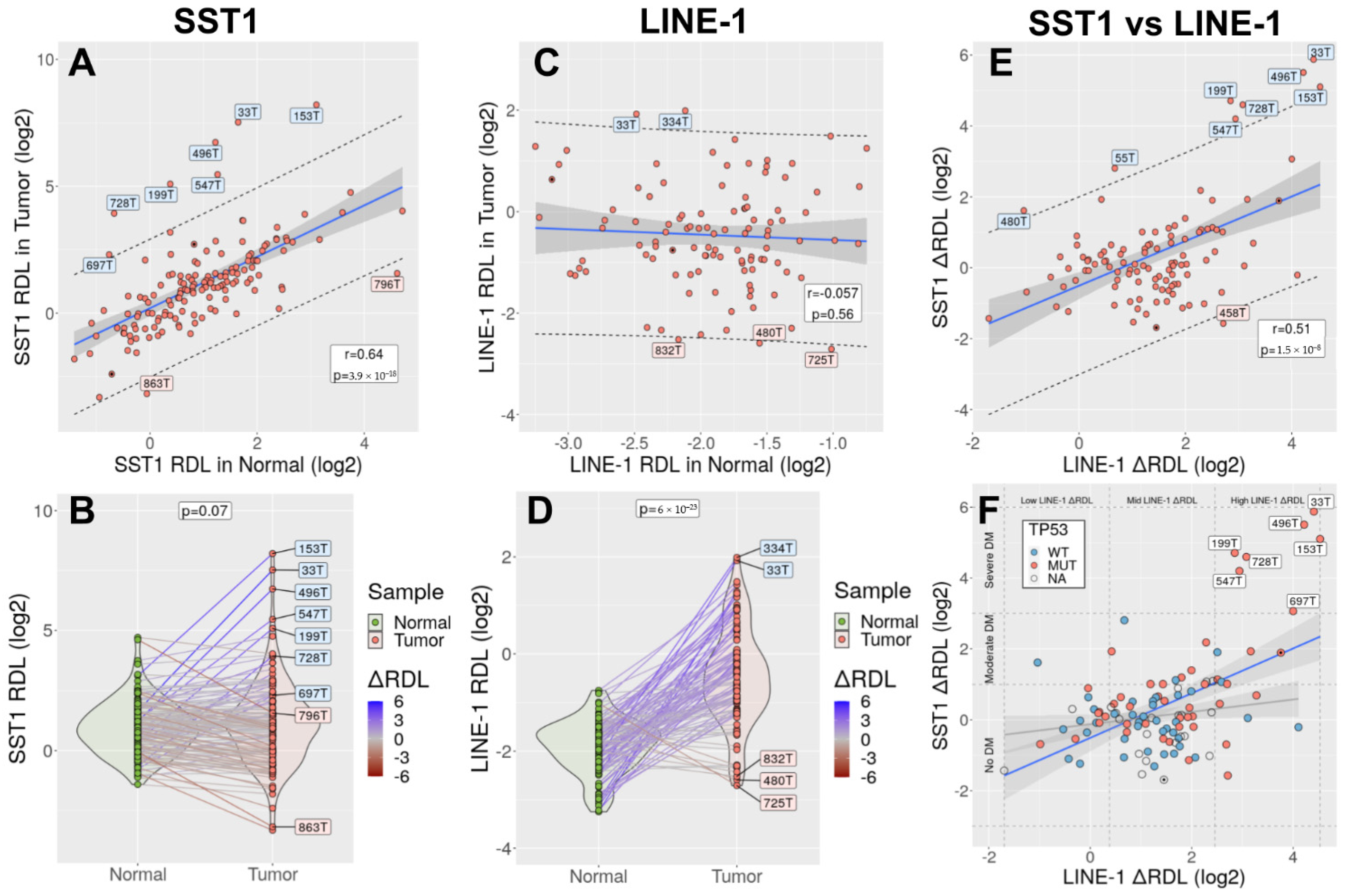
3.6. SST1 Demethylation Associates with LINE-1 Demethylation in Primary CRCs
3.7. SST1 Strong Demethylation Is Associated with TP53 Mutations in CRC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Symer, D.E.; Connelly, C.; Szak, S.T.; Caputo, E.M.; Cost, G.; Parmigiani, G.; Boeke, J.D. Human l1 retrotransposition is associated with genetic instability in vivo. Cell 2002, 110, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Kazazian, H.H., Jr.; Goodier, J.L. LINE drive. Retrotransposition and genome instability. Cell 2002, 110, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.-H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanović, O.; Lister, R. DNA methylation and the preservation of cell identity. Curr. Opin. Genet. Dev. 2017, 46, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.; Moran, S.; Pisano, D.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [Green Version]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA methylation in cancer and aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoal-van Helden, E.G.; van Helden, P.D. Age-related methylation changes in DNA may reflect the proliferative potential of organs. Mutat. Res. 1989, 219, 263–266. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental influences on the epigenome: Exposure-associated DNA methylation in human populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, I.; Rotkopf, H.; Khazaie, K. Bacteria-related changes in host DNA methylation and the risk for CRC. Gut Microbes 2020, 12, 1800898. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Frigola, J.; Song, J.; Stirzaker, C.; Hinshelwood, R.A.; Peinado, M.A.; Clark, S. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006, 38, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Mayor, R.; Casadomé, L.; Azuara, D.; Moreno, V.; Clark, S.J.; Capellà, G.; Peinado, M.A. Long-range epigenetic silencing at 2q14.2 affects most human colorectal cancers and may have application as a non-invasive biomarker of disease. Br. J. Cancer 2009, 100, 1534–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achinger-Kawecka, J.; Taberlay, P.C.; Clark, S.J. Alterations in three-dimensional organization of the cancer genome and epigenome. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bert, S.A.; Robinson, M.D.; Strbenac, D.; Statham, A.L.; Song, J.Z.; Hulf, T.; Sutherland, R.L.; Coolen, M.W.; Stirzaker, C.; Clark, S.J. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 2013, 23, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.E.; van Dijk, C.M.; Tollenaar, R.A.E.M.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.L.; Peters, T.; Khoury, A.; et al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Ou, J.; Hutchinson, L.; Green, M.R. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG island methylator phenotype. Mol. Cell 2014, 55, 904–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, R.W.; Fang, M.; Park, S.M.; Hutchinson, L.; Green, M.R. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. eLife 2014, 3, e02313. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Beisel, C.; Paro, R. Silencing chromatin: Comparing modes and mechanisms. Nat. Rev. Genet. 2011, 12, 123–135. [Google Scholar] [CrossRef]
- Yang, A.S.; Estecio, M.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.-P. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Campan, M.; Long, T.I.; Kim, M.; Woods, C.; Fiala, E.; Ehrlich, M.; Laird, P.W. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005, 33, 6823–6836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Suzuki, I.; Leodolter, A.; Alonso, S.; Horiuchi, S.; Yamashita, K.; Perucho, M. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 2006, 9, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Samuelsson, J.K.; Dumbovic, G.; Polo, C.; Moreta, C.; Alibés, A.; Ruiz-Larroya, T.; Giménez-Bonafé, P.; Alonso, S.; Forcales, S.-V.; Perucho, M. Helicase lymphoid-specific enzyme contributes to the maintenance of methylation of SST1 pericentromeric repeats that are frequently demethylated in colon cancer and associate with genomic damage. Epigenomes 2016, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoraval, D.; Asakawa, J.-I.; Wimmer, K.; Kuick, R.; Lamb, B.; Richardson, B.; Ambros, P.; Glover, T.; Hanash, S. Demethylation of repetitive DNA sequences in neuroblastoma. Genes Chromosom. Cancer 1996, 17, 234–244. [Google Scholar] [CrossRef]
- Nishiyama, R.; Qi, L.; Tsumagari, K.; Weissbecker, K.; Dubeau, L.; Champagne, M.; Sikka, S.; Nagai, H.; Ehrlich, M. A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol. Ther. 2005, 4, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Bobek, M.P.; Kuick, R.; Lamb, B.; Zhu, X.; Narayan, A.; Bourc’his, D.; Viegas-Péquignot, E.; Ehrlich, M.; Hanash, S.M. Whole-genome methylation scan in ICF syndrome: Hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum. Mol. Genet. 2000, 9, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.-L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.-L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Epstein, N.D.; Karlsson, S.; O’Brien, S.; Modi, W.; Moulton, A.; Nienhuis, A.W. A new moderately repetitive DNA sequence family of novel organization. Nucleic Acids Res. 1987, 15, 2327–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Fan, T.; Yan, Q.; Zhu, H.; Fox, S.; Issaq, H.J.; Best, L.; Gangi, L.; Munroe, D.; Muegge, K. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 2004, 32, 5019–5028. [Google Scholar] [CrossRef] [Green Version]
- Dumbovic, G.; Biayna, J.; Banús, J.; Samuelsson, J.; Roth, A.; Diederichs, S.; Alonso, S.; Buschbeck, M.; Perucho, M.; Forcales, S.-V. A novel long non-coding RNA from NBL2 pericentromeric macrosatellite forms a perinucleolar aggregate structure in colon cancer. Nucleic Acids Res. 2018, 46, 5504–5524. [Google Scholar] [CrossRef] [PubMed]
- Dumbović, G.; Sanjuan, X.; Perucho, M.; Forcales, S.-V. Stimulated emission depletion (STED) super resolution imaging of RNA- and protein-containing domains in fixed cells. Methods 2021, 187, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121, 1–84. [Google Scholar] [CrossRef]
- Yamamoto, T.; Rabinowitz, Z.; Sachs, L. Identification of the chromosomes that control malignancy. Nat. New Biol. 1973, 243, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Swanton, C. The role of aneuploidy in cancer evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a028373. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef]
- Barr, F.A.; Gruneberg, U. Cytokinesis: Placing and making the final cut. Cell 2007, 131, 847–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paim, L.M.G.; Fitzharris, G. Tetraploidy causes chromosomal instability in acentriolar mouse embryos. Nat. Commun. 2019, 10, 4834. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.L. Tetraploidy and tumor development. Cancer Cell 2005, 8, 353–354. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Davoli, T.; de Lange, T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; Roos, J.A.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.R.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, J.K.; Alonso, S.; Yamamoto, F.; Perucho, M. DNA fingerprinting techniques for the analysis of genetic and epigenetic alterations in colorectal cancer. Mutat. Res. Mol. Mech. Mutagen. 2010, 693, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Lucito, R.; Healy, J.; Alexander, J.; Reiner, A.; Esposito, D.; Chi, M.; Rodgers, L.; Brady, A.; Sebat, J.; Troge, J.; et al. Representational oligonucleotide microarray analysis: A high-resolution method to detect genome copy number variation. Genome Res. 2003, 13, 2291–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostlander, A.; Meijer, G.; Ylstra, B. Microarray-based comparative genomic hybridization and its applications in human genetics. Clin. Genet. 2004, 66, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Sundström, M.; Göransson Kultima, H.; Botling, J.; Micke, P.; Birgisson, H.; Glimelius, B.; Isaksson, A. Allele-specific copy number analysis of tumor samples with aneuploidy and tumor heterogeneity. Genome Biol. 2011, 12, R108. [Google Scholar] [CrossRef]
- Feber, A.; Guilhamon, P.; Lechner, M.; Fenton, T.; Wilson, G.A.; Thirlwell, C.; Morris, T.J.; Flanagan, A.M.; Teschendorff, A.E.; Kelly, J.D.; et al. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. 2014, 15, R30. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. P53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Medical Association. World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. Bull. World Health Organ. 2001, 79, 373–374. [Google Scholar]
- Robin, T.; Capes-Davis, A.; Bairoch, A. CLASTR: The Cellosaurus STR similarity search tool—A precious help for cell line authentication. Int. J. Cancer 2020, 146, 1299–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.M. Animal Cell Culture: Essential Methods; Wiley: Hoboken, NJ, USA, 2011; ISBN 9780470666586. [Google Scholar]
- Baranovskaya, S.; Soto, J.L.; Perucho, M.; Malkhosyan, S.R. Functional significance of concomitant inactivation of hMLH1 and hMSH6 in tumor cells of the microsatellite mutator phenotype. Proc. Natl. Acad. Sci. USA 2001, 98, 15107–15112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bergkessel, M.; Guthrie, C. Colony PCR. Methods Enzymol. 2013, 529, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Alonso, S.; González, B.; Ruiz-Larroya, T.; Domínguez, M.D.; Kato, T.; Matsunaga, A.; Suzuki, K.; Strongin, A.Y.; Gimenez-Bonafe, P.; Perucho, M. Epigenetic inactivation of the extracellular matrix metallopeptidase ADAMTS19 gene and the metastatic spread in colorectal cancer. Clin. Epigenetics 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Wong, C.C.Y.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Feltz, C.J.; Miller, G.E. An asymptotic test for the equality of coefficients of variation from K populations. Stat. Med. 1996, 15, 646–658. [Google Scholar] [CrossRef]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tom, B.H.; Rutzky, L.P.; Jakstys, M.M.; Oyasu, R.; Kaye, C.I.; Kahan, B.D. Human colonic adenocarcinoma cells. I. Establishment and description of a new line. In Vitro 1976, 12, 180–191. [Google Scholar] [CrossRef]
- Provencher, D.M.; Lounis, H.; Champoux, L.; Tétrault, M.; Manderson, E.N.; Wang, J.C.; Eydoux, P.; Savoie, R.; Tonin, P.N.; Mes-Masson, A.M. Characterization of four novel epithelial ovarian cancer cell lines. In Vitro Cell. Dev. Biol. Anim. 2000, 36, 357–361. [Google Scholar] [CrossRef]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aylon, Y.; Oren, M. P53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef]
- Woerner, S.M.; Benner, A.; Sutter, C.; Schiller, M.; Yuan, Y.P.; Keller, G.; Bork, P.; Doeberitz, M.V.K.; Gebert, J.F.; Yuan, Y. Pathogenesis of DNA repair-deficient cancers: A statistical meta-analysis of putative real common target genes. Oncogene 2003, 22, 2226–2235. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Coquelle, A.; Vivet, S.; Vitale, I.; Kauffmann, A.; Dessen, P.; Pequignot, M.O.; Casares, N.; Valent, A.; Mouhamad, S.; et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006, 25, 2584–2595. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M.; Sanchez, C.; Shao, C.; Nishiyama, R.; Kehrl, J.; Kuick, R.; Kubota, T.; Hanash, S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 2008, 41, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.-A.; Fraga, M.; Morales, C.; Moreno, V.; Esteller, M.; Capella, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpf, A.R.; Matsui, S.-I. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [Green Version]
- Zeimet, A.G.; Fiegl, H.; Goebel, G.; Kopp, F.; Allasia, C.; Reimer, D.; Steppan, I.; Mueller-Holzner, E.; Ehrlich, M.; Marth, C. DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol. Oncol. 2011, 121, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Not Demethylated SST1 ΔRDL < 1.0 | Moderately Demethylated SST1 ΔRDL > 1.0 & < 3.0 | Strongly Demethylated SST1 ΔRDL > 3.0 | Fisher’s Test p-Value |
|---|---|---|---|---|
| Gender | Women (n = 59) | Women (n = 6) | Women (n = 2) | p1 = 0.19 |
| Men (n = 64) | Men (n = 12) | Men (n = 5) | p2 = 0.46 | |
| Age | <66 y/o (n = 57) | <66 y/o (n = 10) | <66 y/o (n = 5) | p1 = 0.27 |
| >66 y/o (n = 66) | >66 y/o (n = 8) | >66 y/o (n = 2) | p2 = 0.27 | |
| Ethnic | Caucasian (n = 94) | Caucasian (n = 9) | Caucasian (n = 3) | p1 = 0.05 |
| Afr.Am. (n = 18) | Afr.Am. (n = 6) | Afr.Am. (n = 1) | p2 = 0.58 | |
| Tumor Location | Proximal (n = 69) | Proximal (n = 10) | Proximal (n = 2) | p1 = 0.38 |
| Distal (n = 50) | Distal (n = 8) | Distal (n = 5) | p2 = 0.24 | |
| Dukes’ stage | IS-A-B (n = 52) | IS-A-B (n = 9) | IS-A-B (n = 4) | p1 = 0.39 |
| C-D-M (n = 71) | C-D-M (n = 9) | C-D-M (n = 3) | p2 = 0.70 | |
| MSI status | MSS (n = 102) | MSS (n = 17) | MSS (n = 6) | p1 = 0.37 |
| MSI (n = 21) | MSI (n = 1) | MSI (n = 1) | p2 = 1.00 | |
| TP53 | WT (n = 44) | WT (n = 5) | WT (n = 0) | p1 = 0.0037 ** |
| MUT (n = 32) | MUT (n = 11) | MUT (n = 7) | p2 = 0.012 * | |
| KRAS | WT (n = 75) | WT (n = 12) | WT (n = 7) | p1 = 0.18 |
| MUT (n = 48) | MUT (n = 6) | MUT (n = 0) | p2 = 0.048 * | |
| BRAF | WT (n = 112) | WT (n = 16) | WT (n = 4) | p1 = 0.25 |
| MUT (n = 10) | MUT (n = 2) | MUT (n = 2) | p2 = 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, B.; Navarro-Jiménez, M.; Alonso-De Gennaro, M.J.; Jansen, S.M.; Granada, I.; Perucho, M.; Alonso, S. Somatic Hypomethylation of Pericentromeric SST1 Repeats and Tetraploidization in Human Colorectal Cancer Cells. Cancers 2021, 13, 5353. https://doi.org/10.3390/cancers13215353
González B, Navarro-Jiménez M, Alonso-De Gennaro MJ, Jansen SM, Granada I, Perucho M, Alonso S. Somatic Hypomethylation of Pericentromeric SST1 Repeats and Tetraploidization in Human Colorectal Cancer Cells. Cancers. 2021; 13(21):5353. https://doi.org/10.3390/cancers13215353
Chicago/Turabian StyleGonzález, Beatriz, Maria Navarro-Jiménez, María José Alonso-De Gennaro, Sanne Marcia Jansen, Isabel Granada, Manuel Perucho, and Sergio Alonso. 2021. "Somatic Hypomethylation of Pericentromeric SST1 Repeats and Tetraploidization in Human Colorectal Cancer Cells" Cancers 13, no. 21: 5353. https://doi.org/10.3390/cancers13215353