
Toll-Like Receptor-4 Antagonist Enhances the Repair of Ultraviolet Radiation-Induced DNA Damage and Augments Anti-Tumor Immune Responses in Mice

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. UVB Light Source and Irradiation of Mice
2.3. RNA Extraction and Real-Time PCR
2.4. Preparation of Tissue Lysates and Western Blot Analysis
2.5. CPD Quantitation by ELISA
2.6. Detection of CPD+ Cells in Skin Sections
2.7. Measurement of Cytokines
2.8. Photocarcinogenesis Study
2.9. Flow Cytometry Analysis
2.10. Statistical Analysis
3. Results
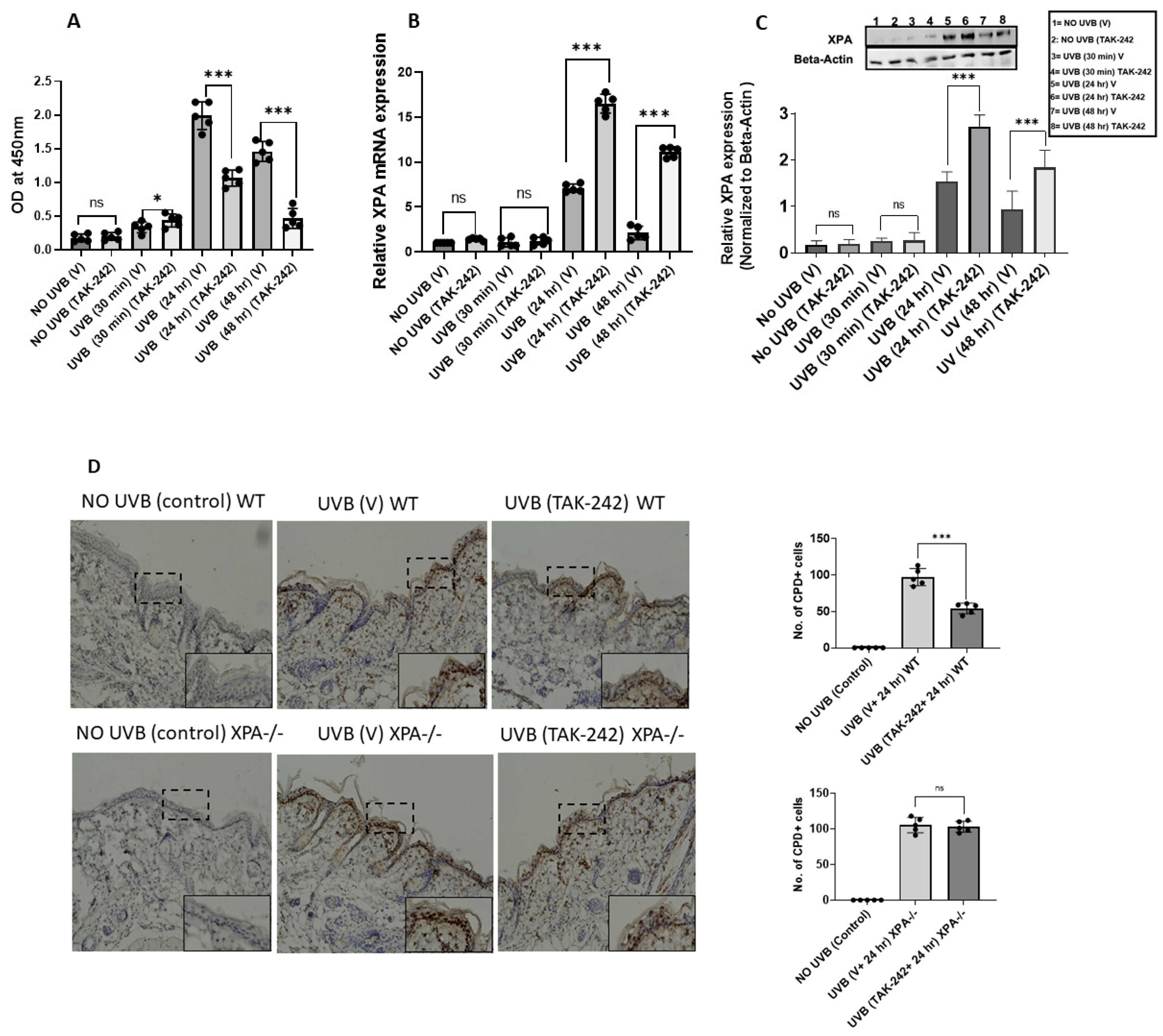
3.1. TAK-242 Treatment Repairs UVB-Induced CPDs
3.2. TLR4 Inhibitor TAK-242 Inhibits UVB-Induced Activation of NLRP3 Inflammasome
3.3. TLR4 Inhibitor TAK-242 Inhibits UVB-Induced Inflammation
3.4. TAK-242 Inhibits UVB-Induced Tumor Development
3.5. TAK-242 Inhibits the Generation of CD4+CD25+ Regulatory T-Cells
3.6. TAK-242 Inhibits UVB-Induced Generation of CD11b+Gr1+ Myeloid Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovach, B.T.; Sams, H.H.; Stasko, T. Systemic strategies for chemoprevention of skin cancers in transplant recipients. Clin. Transpl. 2005, 19, 726–734. [Google Scholar] [CrossRef]
- Fisher, M.S.; Kripke, M.L. Suppressor T lymphocytes control the development of primary skin cancers in ultraviolet-irradiated mice. Science 1982, 216, 1133–1134. [Google Scholar] [CrossRef]
- Kripke, M.L. Antigenicity of murine tumors induced by ultraviolet light. J. Natl. Cancer Inst. 1974, 53, 1333–1336. [Google Scholar] [CrossRef]
- Fisher, M.S.; Kripke, M.L. Further studies on the tumor-specific suppressor cells induced by ultraviolet radiation. J. Immunol. 1978, 121, 1139–1144. [Google Scholar] [PubMed]
- Elmets, C.A.; Bergstresser, P.R.; Tigelaar, R.E.; Wood, P.J.; Streilein, J.W. Analysis of mechanism of unresponsiveness produced by haptens painted on skin exposed to low dose ultraviolet radiation. J. Exp. Med. 1983, 158, 781–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Academy of Dermatology/Milliman. Burden of Skin Disease. 2017. Available online: www.aad.org/BSD (accessed on 4 March 2021).
- Chow, J.; Franz, K.M.; Kagan, J.C. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology 2015, 479, 104–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, G.Y.; Lee, J.W.; Kim, Y.S.; Lee, S.E.; Han, H.D.; Hong, K.J.; Kang, T.H.; Park, Y.M. Interactions between tumor derived proteins and Toll-like receptors. Exp. Mol. Med. 2020, 52, 1926–1935. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Cen, X.; Liu, S.; Cheng, K. The Role of Toll-like Receptor in inflammation and Tumor Immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, N.; Nasti, T.H.; Long, J.A.; Naseemuddin, M.; Lucas, A.P.; Xu, H.; Elmets, C.A. Protective Role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 2008, 68, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Hu, J.; Wang, R.; Han, W.; Zhao, M.; Zhou, G.; Zhang, C.; Zhang, Z. Rapamycin inhibits Toll-like receptor 4- induced pro-oncogenic function in head and neck squamous cell carcinoma. Oncol. Rep. 2014, 31, 2804–2810. [Google Scholar] [CrossRef] [PubMed]
- Pastille, E.; Faβnacht, T.; Adamczyk, A.; Phuong, N.N.T.; Buer, J.; Westerndorf, A.M. Inhibition of TLR4 Signalling Impedes Tumor Growth in Colitis-Associated Colon Cancer. Front. Immunol. 2021, 12, 669747. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E. Toll-Like Receptor Polymorphisms, Inflammatory and Infectious Diseases, Allergies, and Cancer. J. Interferon Cytokines Res. 2013, 33, 467–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, N.; Chaihan, A.; Raithatha, N.; Patel, P.; Khandelwal, R.; Desai, A.; Choxi, Y.; Kapadia, R.; Jain, N. Influence of TLR4 and TLR9 polymorphisms and haplotypes on multiple hrHPV infections and HPV16 copy number in cervical cancer and cervicitis. Microb. Pathog. 2021, 159, 105149. [Google Scholar] [CrossRef]
- Weng, H.; Deng, Y.; Xie, Y.; Liu, H.; Gong, F. Expression and significance of HMGB1, TLR4, and NF-κB p65 in human epidermal tumors. BMC Cancer 2013, 13, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janda, J.; Burkett, N.B.; Mangone, K.B.; Huang, V.; Lewandrowski, C.C.; Albert, D.S.; Petricoin, E.F., 3rd; Calvert, V.S.; Einspahr, J.; Dong, Z.; et al. Resatorvid-based pharmacological antagonism of cutaneous TLR4 blocks UV-induced NF-kappaB and AP-1 signaling in keratinocytes and mouse skin. Photochem. Photobiol. 2016, 92, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Simanyi, E.; Guroji, P.; Tamimi, I.A.; delaRosa, H.J.; Nagar, A.; Nagar, P.; Katiyar, S.K.; Elmets, C.A.; Yusuf, N. Toll-Like Receptor-4 deficiency enhances repair of ultraviolet radiation induced cutaneous DNA damage by nucleotide excision repair mechanism. J. Investig. Dermatol. 2013, 134, 1710–1717. [Google Scholar] [CrossRef] [Green Version]
- Lewis, W.; Simanyi, E.; Li, H.; Thompson, C.A.; Nasti, T.H.; Jaleel, T.; Xu, H.; Yusuf, N. Regulation of ultraviolet radiation induced cutaneous photoimmunosuppression by toll-like receptor-4. Arch. Biochem. Biophys. 2011, 508, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Nasti, T.H.; Rihan, H.M.; Jimenez, H.; Elmets, C.A.; Yusuf, N. Toll like receptor-4 deficiency inhibits ultraviolet radiation-induced tumor development by modulation of immune and inflammatory response. Mol. Carcinog. 2021, 60, 60–70. [Google Scholar] [CrossRef]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, H.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1b production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [Green Version]
- Sand, J.; Haertel, E.; Beidermann, T.; Contassot, E.; Reichmann, E.; French, L.E.; Werner, S.; Beer, H. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine kerationocytes. Cell Death Dis. 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Athar, M.; Elmets, C.A.; Afaq, F. Fisetin inhibits UVB-induced cutaneous inflammation and activation of PI3K/AKT/NF κB signaling pathways in SK-1 hairless mice. Photochem. Photobiol. 2015, 91, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Mangone, K.B.; Burkett, N.B.; Tahsin, S.; Myrdal, P.B.; Aodah, A.; Ho, B.; Janda, J.; McComas, M.; Saboda, K.; Roe, D.J.; et al. Pharmocological TLR4 Antagonism using Topical Resatorvid Blocks Solar UV- Induced Skin Tumorigenesis in SKH-Mice. Cancer Prev. Res. 2018, 11, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, J.; Beura, L.K.; Bobr, A.; Astry, B.; Chicoine, B.; Kashem, S.W.; Welty, N.E.; Igyarto, B.Z.; Wejeyeyesinghe, S.; Thompson, E.A.; et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat. Immunol. 2016, 17, 414–421. [Google Scholar] [CrossRef]
- Ciążyńska, M.; Bednarski, I.A.; Wódz, K.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 inflammasome as a new approach to skin carcinogenesis. Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk with NLRP3 inflammasome and Complement Signalling Pathways in Alzheimers Disease. Front. Immunol. 2020, 23, 724. [Google Scholar] [CrossRef]
- Bashir, M.M.; Sharma, M.R.; Werth, V.P. UVB and pro-inflammatory cytokines synergistically activate TNF-α production in keratinocytes through enhanced gene transcription. J. Investig. Dermatol. 2009, 129, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Nasti, T.H.; Iqbal, O.; Tamimi, I.A.; Geise, J.T.; Katiyar, S.K.; Yusuf, N. Differential roles of T-cell subsets in regulation of ultraviolet radiation induced cutaneous photocarcinogenesis. Photochem. Photobiol. 2011, 87, 387–398. [Google Scholar] [CrossRef]
- Mittal, D.; Saccheri, F.; Venereau, E.; Pusterla, T.; Bianchi, M.E.; Rescigno, M. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010, 29, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Chun, K.S.; Akunda, J.K.; Langenbach, R. Cyclooxygenase-2 inhibits UVB-induced apoptosis in mouse skin by activating the prostaglandin E2 receptors, EP2 and EP4. Cancer Res. 2007, 67, 2015–2021. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vereki, S.M.; Gerhardt, L.; Krishnamoorthy, M. Immunosuppressive Effects of Myeloid-Derived Suppressor Cells in Cancer and Immunotherapy. Cells 2021, 10, 1170. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezernitchi, A.V.; Vaknin, I.; Daniel, L.C.; Levy, O.; Manaster, E.; Halabi, A.; Pikarsky, E.; Shapira, L.; Baniyash, M. TCRζ down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J. Immunol. 2006, 177, 4763–4772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.; Xu, H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid derived suppressor cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin. Cancer Res. 2007, 13, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Guroji, P.; DeBrot, A.H.; Manapragada, P.P.; Katiyar, S.K.; Elmets, C.A.; Yusuf, N. Loss of INK4a/Arf gene enhances ultraviolet radiation-induced cutaneous tumor development. Exp. Dermatol. 2017, 26, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Tober, K.L.; Burns, E.M.; Schick, J.S.; Riggenbach, J.A.; Mace, T.A.; Bill, M.A.; Young, G.S.; Oberyszyn, T.M.; Lesinski, G.B.; et al. UV light B-mediated inhibition of skin catalase activity promotes Gr-1+CD11b+ myeloid cell expansion. J. Investig. Dermatol. 2012, 132, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, A.; Maeda, A.; Wild, M.K.; Kernebeck, K.; Gross, N.; Aragane, Y.; Beissert, S.; Vestweber, D.; Schwarz, T. Ultraviolet radiation-induced regulatory T cells not only inhibit the induction but can suppress the effector phase of contact hypersensitivity. J. Immunol. 2004, 172, 1036–1043. [Google Scholar] [CrossRef]
- Loser, K.; Apelt, J.; Voskort, M.; Mohaupt, M.; Balkow, S.; Schwarz, T.; Grabbe, S.; Beissert, S. IL-10 Controls Ultraviolet-Induced Carcinogenesis in Mice. J. Immunol. 2007, 179, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoreishi, M.; Dutz, J.P. Tolerance induction by transcutaneous immunization through ultraviolet-irradiated skin is transferable through CD4+CD25+ T regulatory cells and is dependent on host-derived IL-10. J. Immunol. 2006, 176, 2635–2644. [Google Scholar] [CrossRef] [Green Version]
- Shreedhar, V.K.; Pride, M.W.; Sun, Y.; Kripke, M.L.; Strickland, F.M. Origin and characteristics of ultraviolet-B radiation-induced suppressor T lymphocytes. J. Immunol. 1998, 161, 1327–1335. [Google Scholar] [PubMed]
- Kaporis, H.G.; Yassky, E.G.; Lowes, M.A.; Haider, A.S.; Duculan, J.F.; Darabi, K.; Whynot-Ertelt, J.; Khatcherian, A.; Cardinale, I.; Novitskaya, I. Human basal cell carcinoma is associated with Foxp3+ T cells in a Th2 dominant microenvironment. J. Investig. Dermatol. 2007, 127, 2391–2398. [Google Scholar] [CrossRef] [Green Version]
- Rogers, H.W.; Coldiron, B.M. Analysis of skin cancer treatment and costs in the United States Medicare population, 1996–2008. Dermatol. Surg. 2013, 39, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Harris, A.R.; Hinckley, M.R.; Feldman, S.R.; Fleischer, A.B.; Coldiron, B.M. Incidence estimate of nonmelanoma skin cancer in the United States. Arch. Dermatol. 2010, 146, 283–287. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | References |
|---|---|---|
| GAPDH | 5′-AACTTTGGCATTGTGGAAGG-3′ 5′-ACACATTGGGGGTAGGAACA-3′ | [18] |
| XPA | 5′-CAAAGGTGGCTTCATTTTAG-3′ 5′-GGTACATGTCATCTTCTAAG-3′ | [18] |
| NLRP3 | 5′-ATTACCCGCCCGAGAAAGG-3′ 5′-TCGCAGCAAAGATCCACACAG-3′ | [21] |
| Caspase-1 | 5′-GGAAGCAATTTATCAACTCAGTG-3′ 5′-GCCTTGTCCATAGCAGTAATG-3′ | [22] |
| ASC | 5′-CAGCAACACTCCGGTCAG-3′ 5′-AGCTGGCTTTTCGTATATTGTG-3′ | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherwani, M.A.; Abdelgawad, A.; Chung, M.; Ibrahim, S.; Eraslan, M.; Elmets, C.A.; Yusuf, N. Toll-Like Receptor-4 Antagonist Enhances the Repair of Ultraviolet Radiation-Induced DNA Damage and Augments Anti-Tumor Immune Responses in Mice. Cancers 2021, 13, 5406. https://doi.org/10.3390/cancers13215406
Sherwani MA, Abdelgawad A, Chung M, Ibrahim S, Eraslan M, Elmets CA, Yusuf N. Toll-Like Receptor-4 Antagonist Enhances the Repair of Ultraviolet Radiation-Induced DNA Damage and Augments Anti-Tumor Immune Responses in Mice. Cancers. 2021; 13(21):5406. https://doi.org/10.3390/cancers13215406
Chicago/Turabian StyleSherwani, Mohammad Asif, Ahmed Abdelgawad, Minh Chung, Saad Ibrahim, Mualla Eraslan, Craig A. Elmets, and Nabiha Yusuf. 2021. "Toll-Like Receptor-4 Antagonist Enhances the Repair of Ultraviolet Radiation-Induced DNA Damage and Augments Anti-Tumor Immune Responses in Mice" Cancers 13, no. 21: 5406. https://doi.org/10.3390/cancers13215406