1. Introduction
Cutaneous squamous cell carcinoma (cSCC) is the second most common non-melanoma skin cancer (NMSC) after basal cell carcinoma. This malignant form of proliferation of the cutaneous epithelium represents 20 to 50% of all the skin cancers and presents with more mutations compared to other common malignancies. These mutations are mostly caused by the exposure to ultraviolet radiation [
1,
2,
3].
Surgery is still the gold standard for the treatment of cSCC, and chemotherapy with cysplatin, 5-fluorouracil, and taxane is also commonly used [
4]. However, cosmetic limitations strongly motivate the use of non-invasive procedures, such as photodynamic therapy (PDT), which has become paramount to treat this malignancy [
5]. In addition to the good clinical response to PDT and high cure rate of both invasive and in situ SCC (Bowen’s disease), it is also worth mentioning that PDT is being used as a field treatment for the well-known precursor lesion of cSCC named actinic keratosis, with a successful preventive outcome [
5,
6,
7]. Besides, PDT is clinically approved for the treatment of other forms of NMSC, including basal cell carcinoma and other types of cancer, such as oesophageal cancer [
8,
9].
PDT is a non-invasive and localized therapy that exerts minimal or no damage and improved healing capacity in the surrounding healthy tissues [
10,
11]. It consists of a two-step procedure by which the local or systemic administration of a photosensitizer (PS) or its precursor is followed by the irradiation of the lesion with light of a specific wavelength. The interaction between the PS and the light in the presence of molecular oxygen leads to the generation of reactive oxygen species (ROS), which are responsible for the induction of cancer cell death [
6,
12,
13,
14]. One of the two most commonly used topical drugs approved for the treatment of NMSC with PDT is 5-methyl aminolevulinate (MAL), the methyl ester form of the aminolevulinic acid (ALA). Both are precursors of protoporphyrin IX (PpIX), an endogenous PS produced as an intermediate metabolite of the heme biosynthetic pathway [
15,
16,
17]. Even though PDT is an excellent treatment for the indicated applications, sometimes the lesions can recur [
18].
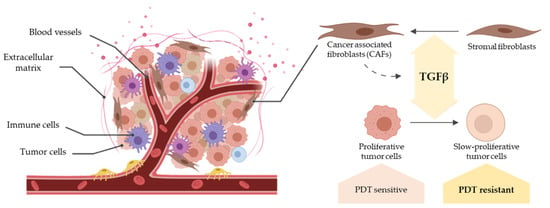
In the last few years, the perception of cancer biology by the scientific community has changed dramatically, no longer focusing exclusively on tumor cells. In this regard, the complex microenvironment around them, known as the tumor microenvironment (TME), has gained importance [
19,
20]. The stromal component of the TME is composed of different cell types accompanied by non-cellular components, such as the extracellular matrix (ECM), as well as by other elements, including blood vessels. Overall, this intricate structure makes carcinomas very heterogeneous tissues. Previous studies have shown that the TME plays a key role in tumor initiation, promotion, progression, and metastasis, being directly related to the development of resistances against different therapies, including chemotherapy [
19,
20,
21,
22]. Within the TME-resident cell populations, cancer-associated fibroblasts (CAFs) stand out because of their wide effects on both the tumor and its stroma. CAFs are activated fibroblasts associated with the enhancement of the aforementioned stages of carcinogenesis and known to secrete several growth factors and cytokines that promote TME remodelling [
19,
23]. They have been shown to contribute to tumor progression through the enhancement of cellular migration and the alteration of tumor cell metabolism. Thus, CAFs have been characterized as an effective prognostic factor in different types of tumors [
20].
CAFs differ from normal fibroblasts in the expression of different proteins that serve as identity markers [
21,
24]. Among these, alpha smooth muscle actin (α-SMA), vimentin, CD10, and fibronectin can be highlighted. α-SMA, which has been associated with the enhancement of tumor progression [
24,
25,
26], is a highly conserved member of the actin family with an important role in cell motility, structure and integrity. Vimentin is an intermediate filament protein commonly used as an epithelial-to-mesenchymal transition biomarker, related to cell migration and maintenance of the structure; therefore, its overexpression is associated with an invasive phenotype [
27,
28]. CD10 is a neutral endopeptidase expressed by both neoplastic and stromal cells and involved in the regulation of cell proliferation and invasion, as well as in the degradation of peptides and cytokines in the TME. Thus, the expression of CD10 is associated with poor clinical outcomes [
29,
30]. Finally, fibronectin is one of the most abundant ECM proteins which has been highly studied due to its role in the modulation of the ECM. Its overexpression has also been associated with tumor progression [
31,
32].
Importantly, CAFs secrete different cytokines to the TME, among which transforming growth factor-β (TGFβ) is a major one [
32]. This ubiquitously expressed cytokine controls several biological processes through the regulation of proliferation, migration, angiogenesis, apoptosis, or ECM remodeling [
33,
34]. When TGFβ is secreted and activated in the ECM, it binds to the type II and type I receptors (TβRII, TβRI), triggering the formation of a receptor complex. These steps are mediated by the phosphorylation of both receptor types, and lead to the phosphorylation of the cytoplasmic proteins Smad2 and 3, which then bind to Smad4. The phosphorylated Smad complex subsequently binds specific DNA sequences (Smad Binding Elements, SBEs), regulating the expression of TGFβ target genes [
34,
35,
36]. In addition, the presence of coreceptors in the cell membrane, such as endoglin, can influence the downstream effects of TGFβ [
36].
Under pathological conditions, such as cancer, the TGFβ pathway is highly upregulated due to increased levels of the active ligand in the TME [
33,
34]. Aside from the CAF-derived TGFβ, cancer cells themselves produce this cytokine in variable levels, helping to further recruit and activate CAFs. Altogether, this leads to a global increase in the production and activation of TGFβ [
34,
35]. In spite of its highly complex role in cancer, it has been described that TGFβ acts as a suppressor in the early stages of tumorigenesis and as a promoter in later phases, revealing the stage-specific effects of this cytokine [
33]. More specifically, many studies are focused on TGFβ1 as it is the most abundant isoform [
35]. In particular, studies in cSCC have reported an overexpression of TGFβ1 in the non-malignant tissue surrounding the tumor, followed by a decrease in expression after surgical resection [
33,
35], supporting the previously mentioned tumor-promoting effect. In contrast, a TGFβ1-dependent cell cycle arrest at the G1 phase has been described in both normal epithelium and tumor tissue, potentially supporting a role in tumor suppression [
33,
34,
35]. Interestingly, however, this cell cycle arrest promoted by TGFβ1 has been related to drug resistance in different cancers, including cSCC, as a consequence of the direct repression of specific genes related to cell cycle progression by Smad2/3 proteins [
22,
37].
In the case of resistance to PDT, CAF-derived TGFβ1 has been identified as an extrinsic factor in different cancers, including cSCC. In the first place, TGFβ1 induces CAFs to produce fibrotic components modifying the composition of the ECM. These desmoplastic matrices can obstruct the PS delivery and therefore impair PDT [
32,
35]. Secondly, TGFβ1 promotes epithelium-to-mesenchymal transition (EMT) and the acquisition of stem cell-like phenotypes, further contributing to create a fibrotic ECM and to the resistance to therapy. Finally, as previously stated, TGFβ1 produced by CAFs leads to resistance through the cell cycle arrest at G1 phase. Moreover, arrested cells that engage a quiescent state have been identified as drivers of resistance and recurrence in SCC tumors [
35,
37].
Taking all that into account, the objective of this work is to evaluate the implications of TGFβ1 produced by CAFs in the response of cSCC to PDT, with a particular focus on the molecular mechanisms that can drive resistance to this therapy.
2. Materials and Methods
2.1. Cell Culture
Two established human cell lines from cSCC, A431 and SCC13, were used [
38]. A431 corresponds to a poorly differentiated vulvar tumour, while SCC13 corresponds to a moderately differentiated facial tumour.
Fibroblast primary cultures were obtained from adult patients with in situ SCC (Bowen disease) (T165A and T213A) and invasive SCC (T205A and T223A) in the case of CAFs, and from healthy donors in the case of controls (C1 and C3 were of child origin and C2 was isolated from a healthy adult). Cc, a dermal fibroblast culture from a healthy adult, was purchased from Innoprot (Innovative Technologies in Biological Systems, Derio, Spain).
Human fibroblasts used in the experiments were isolated from 2-mm biopsies from patients with cSCC, with the approval of the Ethics Committee of the Hospital Ramón y Cajal (Madrid, Spain). Once obtained, biopsies were conserved in saline serum for a short time until cultured. For the isolation of fibroblasts, a protocol modified from previously published studies [
38,
39] was used. Briefly, biopsies were chopped in a Petri dish with 1 mL of dispase II (Sigma, St. Louis, MO, USA) after washing with 1:1 ethanol (Panreac, Barcelona, Spain) and PBS (Phosphate Buffered Saline, Thermo Scientific Inc., HyClone, Rockford, IL, USA). Afterwards, tissues were incubated with 5 mL of dispase II overnight at 4 °C and centrifuged 10 min at 390 g. The pellet was washed with PBS (Thermo Fisher Scientific Inc., Rockford, IL, USA) and centrifuged two additional times, followed by the transference to a flask with DMEM (Dulbecco’s modified Eagle’s medium, high glucose) supplemented with 10% (
v/
v) fetal bovine serum (FBS) and 1% antibiotics (penicillin, 100 units/mL; streptomycin 100 mg/mL), all from Thermo Fisher Scientific Inc. (Rockford, IL, USA). When cells had reached 70% confluence, fibroblasts were separated from keratinocytes by differential detachment using 0.05% trypsin (Thermo Fisher Scientific Inc., Rockford, IL, USA).
Cell lines and primary fibroblasts were grown in DMEM supplemented with 10% (v/v) FBS and 1% antibiotics (penicillin, 100 units/mL; streptomycin 100 mg/mL). Cell cultures were performed under standard conditions of 5% CO2, 95% humidity, and at 37 °C. Consecutive passages of cSCC cell lines were achieved by treatment with 1 mM of EDTA/0.25% trypsin (w/v), while 1 mM of EDTA/0.05% trypsin (w/v) was used for fibroblasts (both trypsin solutions from Thermo Fisher Scientific Inc., Rockford, IL, USA), followed by centrifugation at 480 g for 5 min at room temperature.
2.2. Western Blot
For Western blot (WB), fibroblasts or cSCC cells were seeded in 6-well plates and protein extracts were obtained using RIPA buffer (BioRad, Hercules, CA, USA) mixed with Triton X-100 (pH 7.4, Bioworld, Dublin, OH, USA), phosphatase inhibitors (PhosSTOP EASYpack, Roche, Mannheim, Germany), and protease inhibitors (complete ULTRA tablets Mini EDTA-free EASYpack, Roche, Mannheim, Germany). Afterwards, protein concentration was determined with the BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, IL, USA). Protein extracts from fibroblast were diluted in Laemmli buffer mixed with β-mercaptoethanol (Bio-Rad, Hercules, CA, USA) and heated for 10 min at 98 °C. Afterwards, extracts were centrifuged for 10 min at 17,968 g at 4 °C. Electrophoresis was performed by using acrylamide/bis-acrylamide gels in denaturing conditions (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA), using a Transblot Turbo system (Bio-Rad, Hercules, CA, USA). Membranes were blocked in skimmed milk in 0.1% TBS-Tween-20, then incubated with primary antibodies against α-SMA (Abcam, Cambridge, UK), vimentin (Abcam), CD10 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), and α-tubulin (Cell Signalling Tecnology, Inc., Danvers, MA, USA) for fibroblasts extracts, as well as HO-1 (Cell Signalling) and vinculin (Sigma-Aldrich, St. Louis, MO, USA) for cSCC extracts. Afterwards, they were further washed and incubated with their corresponding peroxidase-conjugated secondary antibodies (HRP-Goat anti-rabbit IgG and HRP-Goat anti-mouse IgG, Thermo Fisher, Rockford, IL, USA). Protein bands were visualized by chemiluminescence (ECL Plus Kit, Amersham, Little Chalfont, UK) using the high-resolution ChemiDocTR XRS+ system (Bio-Rad) and digitized using Image Lab version 3.0.1 software (3.0.1. version, Bio-Rad Software, Hercules, CA, USA).
2.3. Immunofluorescence
For indirect immunofluorescence (IF), fibroblasts were grown on glass coverslips until reaching around 70% confluence and then fixed with formaldehyde (Sigma-Aldrich) diluted to 3.7% in PBS at 4 °C (Thermo Fisher Scientific Inc.). Afterwards, cells were washed three times with PBS and permeabilized with 0.5% Triton X-100 in PBS at room temperature. The cells were then incubated with primary antibodies against α-SMA (Abcam), vimentin (Abcam), CD10 (Invitrogen, Thermo Fisher Scientific Inc.) or fibronectin (Abcam) diluted in 0.5% bovine serum albumin (BSA, Sigma-Aldrich) in PBS for 1 h at 37 °C. After three washes, they were incubated with the secondary antibodies AF546 goat anti-rabbit IgG or AF488 goat anti-mouse IgG (Thermo Fisher Scientific Inc.) for 45 min at 37 °C. Afterwards, cells were washed again and incubated with Höechst 33258 (Sigma-Aldrich) for 5 min at 37 °C for nuclear counterstaining. Finally, the coverslips were mounted with ProLong® (Life Technologies, Carlsbad, CA, USA). The slides were observed using an epifluorescence microscope (Olympus BX61, Olympus Corporation, Shinjuku, Tokyo, Japan) equipped with a HBO 100 W mercury lamp and filter sets for fluorescence microscopy, using blue light irradiation (450–490 nm, BP 490 filter, Olympus Corporation, Shinjuku, Tokyo, Japan) for CD10; ultraviolet irradiation (360–370 nm, UG-1 filter, Olympus Corporation, Shinjuku, Tokyo, Japan) for nuclei; and green light irradiation (570–590 nm, DM 590 filter, Olympus Corporation, Shinjuku, Tokyo, Japan) for α-SMA, vimentin, and fibronectin.
For the quantification of the degree of alignment of fibronectin fibres, IF images were processed using the ImageJ software (version 1.8.0, ImageJ, Bethesda, MD, USA). Briefly, after black to white inversion, the images were convolved and the area of the fluorescent signal was measured. A minimum of four images per condition were included in the analysis.
2.4. Measurement of Secreted TGFβ1
To evaluate the production of TGFβ1 by cells in culture, the Quantikine® ELISA Human TGFβ1 Immunoassay (R&D Systems, Minneapolis, MN, USA) kit was used. To obtain the samples, cells were seeded in 24-well plates. When they reached 80% confluence, medium was switched to 250 µL of phenol red-free DMEM (Thermo Fisher Scientific Inc., HyClone, Rockford, IL, USA) supplemented with 1% FBS and 1% antibiotics. After 24 h, supernatants were harvested, centrifuged at 480 g for 5 min and were stored at −20 °C until ulterior analyses. Before performing the quantification assay, latent TGFβ1 present in the supernatants was activated for its detection by incubating the samples with 1 M of hydrochloric acid, followed by neutralization by adding a solution containing 1.2 M of sodium hydroxide and 0.5 M of HEPES. In parallel, cells remaining in the wells were counted using a TC20TM automated cell counter (BioRad) to normalize the TGFβ1 measurements.
2.5. Treatment with Conditioned Medium and Exogenous TGFβ1
CM was obtained from fibroblasts seeded and cultured under standard conditions in 75 cm2 flasks. When 80% confluence was reached, medium was changed to DMEM with 1% FBS and 1% antibiotics. After 24 h of incubation, the CM was collected for the treatment of tumoral cells. For the treatment with TGFβ1 administered exogenously, lyophilized recombinant human TGFβ1 (Preprotech) was reconstituted in 10 mM of citric acid at a concentration of 1 μg/mL, finally adjusting the pH at 7.4. From this stock, the different concentrations of TGFβ1 required for the treatments were prepared in DMEM with 1% FBS and 1% antibiotics. All TGFβ1 treatments (endogenous and exogenous) were performed during 48 h.
2.6. Spheroid Cultures
Spheroid three-dimensional cultures were prepared using A431 and SCC13 cells in a specific medium made of 1:1 DMEM and F12 (F-12 Nutrient mixture, Ham, Gibco, Thermo Scientific Inc., Rockford, IL, USA), 2% B27 serum free supplement (Gibco), 20 ng/mL of EGF (Sigma-Aldrich), 0.4% BSA, and 4 μg/mL of insulin (Gibco). Before seeding the cells, multi-well plates were precoated with 1.2% poly-HEMA (2-hydroxyethyl methacrylate, Sigma-Aldrich) and incubated overnight at room temperature to create a film so that the cells could not adhere to the surface. Then, the cells were plated at a density of 40,000 cells/mL, taking 6 days to form the spheroids.
2.7. Photodynamic Therapy and Cell Viability Assessment
For the administration of PDT, 10 mM of stock Methyl-aminolevulinate (MAL) (Sigma-Aldrich) was prepared in deionized sterile water. SCC cells were seeded and grown in culture conditions up to 60–70% confluence. Then, cells were incubated with 0.5 mM MAL for 5 h in DMEM culture medium without FBS and subsequently irradiated with red light doses ranging between 0.6 and 12.2 J/cm
2. A red-light emitting diode source (WP7143 SURC/E Kingsbright, Angels, CA, USA) with an irradiation intensity of 6.2 mW/cm
2 and an emission peak at λ = 634 ± 20 nm was used. To minimize light refraction, cells were irradiated from the bottom of the plates. After irradiation, cells were incubated for 24 h in normal conditions before evaluation. This treatment conditions were selected according to previous works performed in our research group as capable to induce cell death in SCC13 and in A431 [
10,
40].
For PDT resistance studies, A431 and SCC13 cell lines were seeded as described. Then, 24 h after seeding, culture medium was removed, and cells were incubated during 48 h prior to PDT with CM or fresh culture medium containing exogenous TGFβ1, or DMEM with 1% FBS and 1% antibiotics for MAL-PDT. In the case of CM and exogenous TGFβ1 treatments, the concentration of FBS was maintained at 1% throughout the administration of PDT to avoid interferences with the TGFβ1 present in it. Following the pre-treatment, PDT was administered by incubating the cells for 5 h with 0.5 mM of MAL diluted in the corresponding media and subsequently irradiating with the red light doses indicated above. After PDT, the medium was removed and the corresponding media were added to each condition.
To block TGFβ1 signalling, the compounds used were the SB525334 inhibitor of TGFβ1 receptor I (Sigma-Aldrich, kindly provided by Dr. María Luisa Botella, CIB-CSIC, Madrid, Spain) and a TGFβ-neutralizing antibody (R&D Systems). For the administration of SB525334, a 100 mM of stock prepared in DMSO was diluted in DMEM with 1% FBS to reach a final concentration of 1 µM that was added to the cells 15 min prior to the treatment with CM (according to previous works [
41]) and it was maintained all along the experiment, performed in the same conditions as indicated above. For the administration of the neutralizing antibody, 1 mg/mL of stock prepared in sterile PBS was diluted in DMEM with 1% FBS. The anti-TGFβ was used at 3 µg/mL, added to the cells 48 h prior to MAL-PDT, and was maintained all along the experiment, as indicated above. Treatment conditions were selected according to previous works [
42].
The toxicity of MAL-PDT in cells was assessed 24 h after the phototreatments. The MTT assay was used for bidimensional cultures. To this end, a stock solution of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma-Aldrich) in PBS (1 mg/mL) was prepared and diluted in complete medium to obtain a final concentration of 50 μg/mL to be added to the cells and incubated for 3 h at 37 °C. After removing the solution, DMSO was added to dissolve the formazan crystals formed. The absorbance was measured at 542 nm by using the plate reader SpectraFluor, Tecan (Zürich, Switzerland). The results were represented as percentage of cell survival.
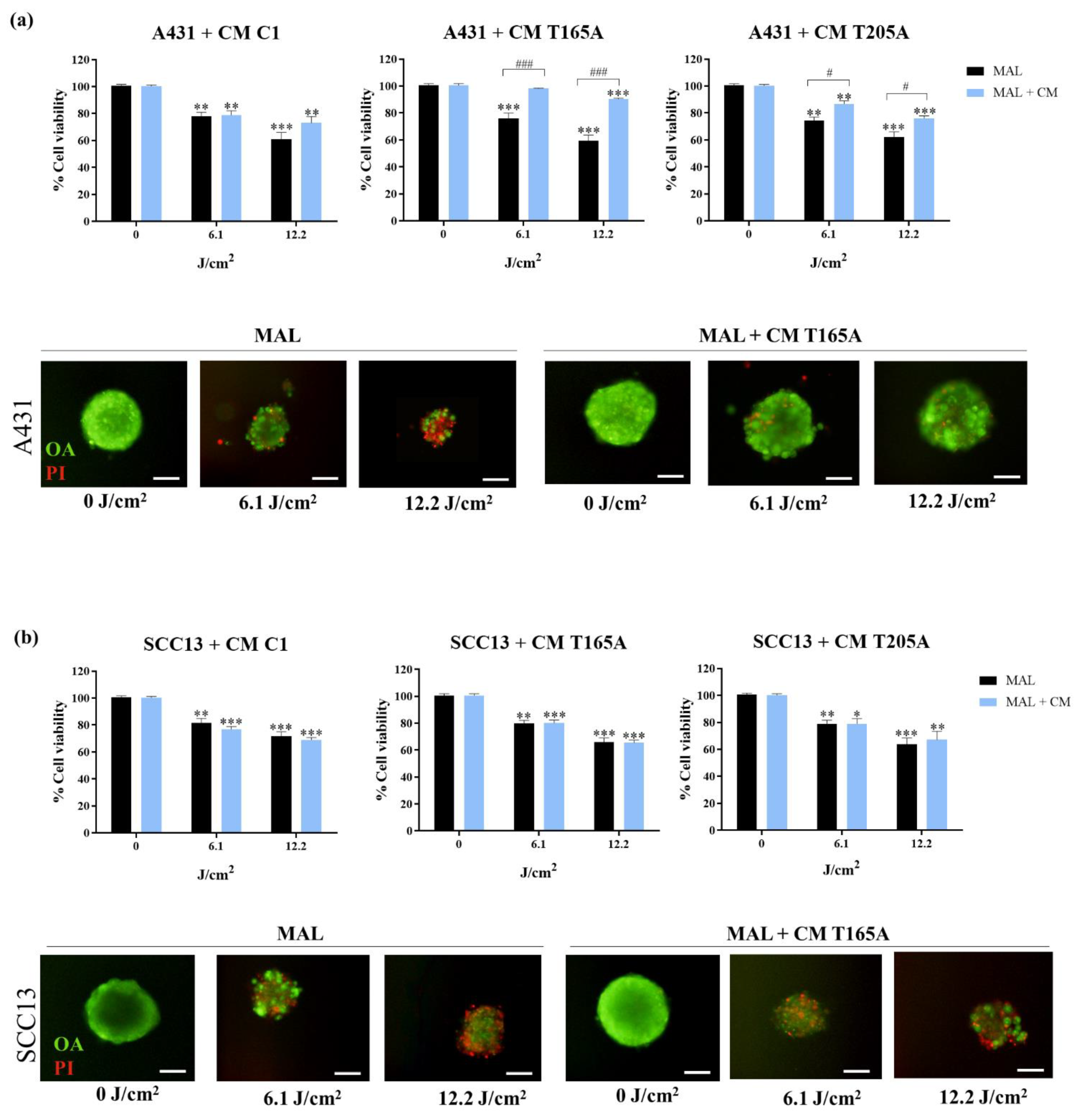
In the case of spheroids, they were preincubated with CM for 48 h and subsequently exposed to MAL-PDT. The toxicity of MAL-PDT was determined 24 h after irradiation by propidium iodide-orange acridine assay (PI/OA). The PI red signal served to estimate the % of cells that had lost membrane integrity (dead cells) over the total number of cells forming the spheroid (both alive and dead, all of them stained in green with OA). The fluorescent signal of the compounds was observed using the fluorescence microscope with blue light irradiation (450–490 nm, BP 490 filter) for OA and green light irradiation (570–590 nm, DM 590 filter) for PI. The images obtained were analysed using Image J and represented as an estimation of cell survival rates.
The migration assay was performed after the exposition of spheroids to MAL-PDT in both cases, with or without pre-treatment with CM. Spheroids with their respective medium were transferred to a 12-well plate with coverslips to allow attachment of living cells to the well. Photographs of the development of the spheroids were taken at 0 h, 4 h, and 24 h by using contrast phase microscopy. The cells that survive MAL-PDT are expected to attach to the surface and migrate, while dead cells will remain in suspension.
The diameter of the spheroids was measured using Image J and represented with the GraphPad Prism software (version 6.05, GraphPad Software Inc., San Diego, CA, USA).
2.8. Cell Proliferation Assessment
A431 and SCC13 cells were grown on glass coverslips until reaching around 70% confluence and then fixed with 3.7% formaldehide in PBS. Afterwards, cells were washed three times with PBS, permeabilized with 0.5% Triton X-100-PBS at room temperature, and incubated with Höechst for 5 min. Finally, the coverslips were mounted with ProLong®. The fluorescent signal of the nuclei was observed under the fluorescence microscope by using ultraviolet irradiation (360–370 nm, UG-1 filter, Olympus Corporation, Shinjuku, Tokyo, Japan). Dividing and non-dividing cells were counted to estimate the mitotic index and the mean +/− SEM was represented.
2.9. ROS Production
The production of ROS was evaluated by flow cytometry in both cSCC cell lines. To this end, cells were incubated with 6 µM of dihydro-fluorescein diacetate (DFH-DA) (Abcam) for 1 h, then trypsinized and centrifuged over 5 min at 600 g. The pellet was then fixed with 3.7% formaldehyde for 15 min at room temperature. The fixative solution was then removed after centrifugation at 600 g for 10 min. The pellets were stored at 4 °C until the evaluation by flow cytometry (Cytomics FC500, 2 lasers, Beckman Coulter) (490–530 nm). The fluorescence intensity was determined in 10,000 cells of each population.
2.10. Statistical Analyses
All the experiments were repeated at least three times. The statistical analysis and data representation were carried out using the GraphPad Prism software (version 6.05, GraphPad Software Inc., San Diego, CA, USA). A one-way ANOVA test was performed to compare the results obtained at different treatment conditions in comparison with one control condition. A t-test was used for the comparisons between two groups. Statistical significance was set at p < 0.05.
4. Discussion
The contribution of CAFs to tumor progression and aggressiveness through the secretion of a variety of factors to the TME is well known [
19]. Among all these factors, TGFβ has been demonstrated to induce resistance to different therapies [
19,
20,
21]. In this sense, CAFs are known to be a major source of this growth factor [
23,
31]. More specifically, studies carried out in mouse models with cSCC have proven that TGFβ1 mediates tumor resistance to different chemotherapeutic agents [
22,
35]. However, no studies have yet addressed this question in the field of PDT, which is becoming a very common therapeutic strategy for the treatment of cSCC given its aforementioned advantages [
5,
6]. Taking all this into account, our aim was to uncover the implications of CAF-derived TGFβ1 in the modulation of the response of cSCC to PDT and in the development of resistance mechanisms.
As other authors have reported, the most accurate method to identify and distinguish CAFs from normal fibroblasts is the characteristic pattern of expression of specific markers related to their activity and effects in tumor resistance [
24,
25,
26]. On that account, and based on the implications of TGFβ1 in cSCC resistance to other therapies [
22], we prioritized the analysis of the endogenous levels of secretion of this cytokine for the selection of T165A and T205A as the most representative CAF cultures, and C1 as a control. On the other hand, despite α-SMA being one of the best known CAF markers, our results were controversial, generally showing lower expression levels of this marker in the majority of CAFs isolated from patients. However, some studies have revealed that the expression of α-SMA may vary among different CAF populations [
24], pointing out that its expression levels can show a wide variation in activated fibroblasts that still behave as CAFs. Related to the other markers, the results obtained along the characterization were consistent, since the overexpression of vimentin and CD10 and the parallel alignment of fibronectin fibers are all well-known features of CAFs [
26,
27,
30].
The involvement of CAFs in the resistance to different chemotherapeutic agents has been demonstrated by other authors in prostate, pancreatic, breast, and gastric cancers, among others [
41]. Although previous work has also been carried out in SCC [
22], our study addresses this question for the first time in regard to PDT. Our findings indicate that CAFs can mediate resistance to PDT in cSCC. Furthermore, this resistance was necessarily induced by secreted factors, given that our experimental approach was restricted to the use of CAF-derived CM. Interestingly, the resistance to PDT was only observed in the A431 cell line, but not in SCC13. Based on previous literature [
22,
34,
36], we demonstrated that the TGFβ1 component of the CAF-derived CM was responsible for the induction of resistance to PDT in A431 cells, as this effect was abrogated by inhibiting TGFβ signalling through the use of the SB525334 TGFβ1 receptor I inhibitor, which has been extensively used in previous studies [
41]. Complementary assays using recombinant TGFβ1 recapitulated the decrease in PDT toxicity in A431 cells and confirmed that this cytokine is capable of driving resistance to MAL-PDT in cSCC cells.
Our results have shown that the TGFβ1 secreted by CAFs is capable of inducing resistance to PDT in both bidimensional and three-dimensional A431 cell cultures. In this sense, migration assays using A431 spheroids showed that CAF-derived CM could also promote EMT, since increased explant expansion was observed from A431 spheroids treated with CAF-derived CM in combination with PDT compared to PDT alone. This results are in line with existing literature [
44] and a deeper characterization of this aspect will need to be addressed in future studies. Overall, these data broaden the implications of our findings, as the use of multicellular spheroids is a powerful tool that more accurately mimics the in vivo events.
The fact that the effect of resistance to PDT was observed in A431 but not in SCC13 cells poses the question of intrinsic differences between the human-originated cSCC cell lines used in this study, even though both are widely used as in vitro models for the study of this type of cancer [
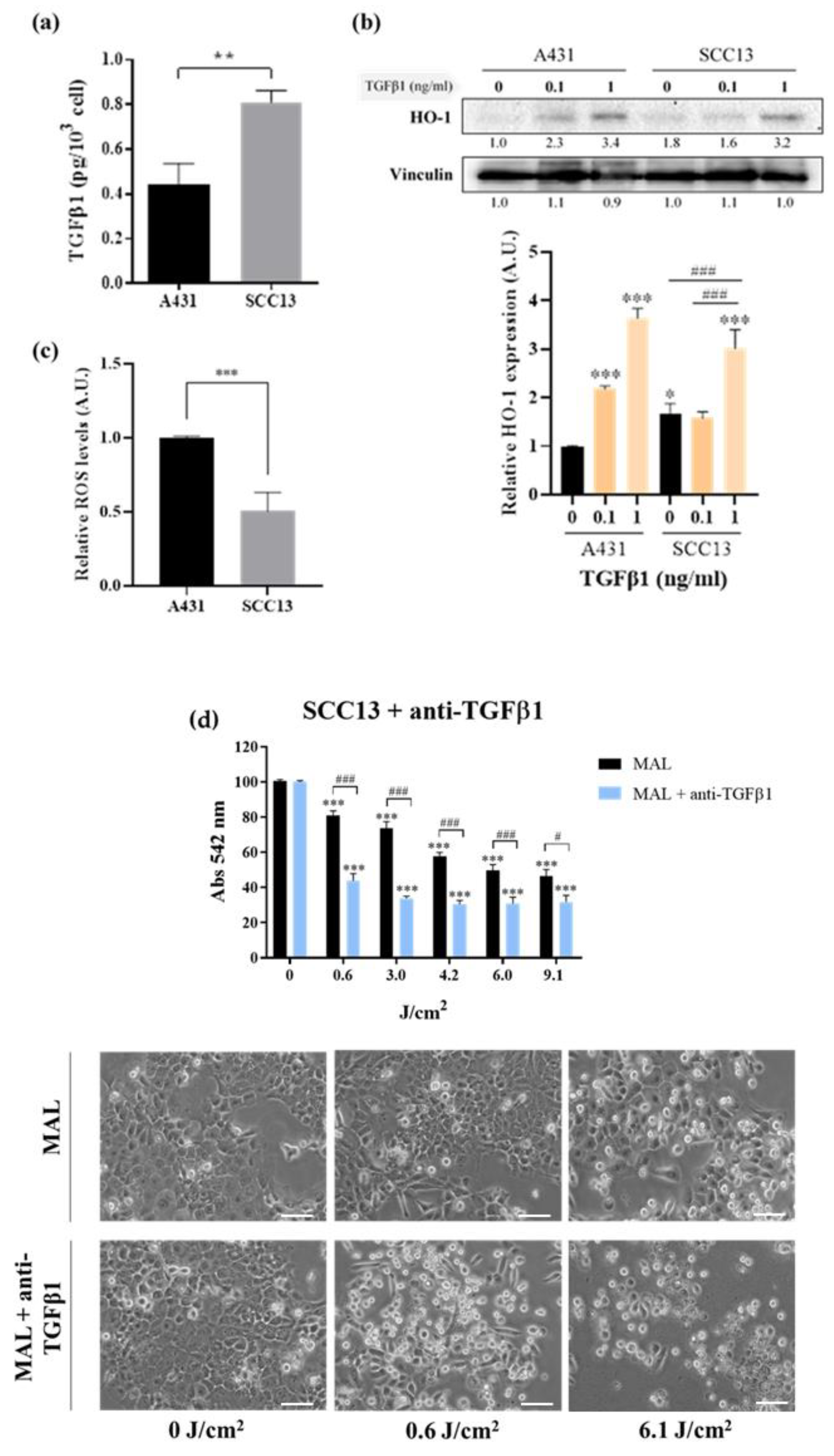
37]. In fact, we found significantly different levels of endogenous TGFβ1 secreted by these two cSCC cell lines. In line with this, several authors have reported an association of the prodution of TGFβ1 with the response to PDT. More specifically, this cytokine induces an overexpression of HO-1 through the upregulation of P38 MAPK and PI3K pathways [
45,
46]. In agreement with this, our results indicate that higher secretion levels of endogenous TGFβ1 correlate with incresed expression of HO-1 in SCC13 cells.
In this sense, other authors have reported that dysregulations affecting the components of the heme synthesis pathway can compromise the optimal functioning of PDT [
47], which are the ultimate effectors of PDT [
13]. In this regard, the reduced basal ROS levels found in SCC13 cells may contribute to the intrinsic relative resistance to PDT displayed by this cell line and limit the susceptibility to develop increased resistance to PDT in response to exogenous TGFβ1. Accordingly, some authors have evidenced that higher secretion levels of endogenous TGFβ1 can reduce the sensitivity to exogenous TGFβ1, probably due to the saturation of the pathway by the autocrine signalling [
44,
48,
49]. Hence, the high secretion levels of TGFβ1 found in SCC13 cells could further explain the lack of resistance to PDT observed in this cell line in the presence of TGFβ1. In contrast, the relatively higher basal ROS levels found in A431 cells are in line with the reduced endogenous TGFβ1 production and comparatively low expression of HO-1. Altogether, these results help to interpret the molecular mechanisms driving the higher sensitivity of A431 cells to PDT as well as the susceptibility of this cell line to develop resistance to PDT in the presence of exogenous TGFβ1.
The development of resistance to therapies in response to TGFβ1 has been tightly related by other authors to the slowing down of cell cycle progression [
22,
36,
44,
48,
50]. In agreement with this, our data suggest that A431 cells undergo a decrease in proliferation upon the exposure to TGFβ1, evidenced by a shift into more flattened morphologies and the reduction in the mitotic index. Indeed, the cell cycle arrest has been reported as an effect of TGFβ1 and linked to resistance to therapies in previous studies [
22,
35,
51]. Taken together, these data support the fact that TGFβ1 is responsible for the resistance to PDT caused by CAFs in A431 cells, likely acting through the induction of cell quiescence. On the other hand, neither changes in the mitotic index nor resistance to PDT were triggered in SCC13 cells by the CM from CAFs, and the threshold concentration of recombinant TGFβ1 needed to decrease cell proliferation in this cell line was shown to be higher than in A431 cells.
The fact that exogenous TGFβ1, even at relatively high concentrations, is able to reduce cell proliferation in SCC13 cells but does not lead to resistance to PDT may seem controversial. In this regard, our data point to the altered expression of components of the heme synthesis pathway, such as HO-1, as responsible for the intrinsic resistance to PDT of this cell line. This could explain why the expected effects of TGFβ1 in SCC13 are observed at the level of cell proliferation but not in the response to the PDT. Altogether, these results are consistent with a refractory phenotype of SCC13 cells that prevents the acquisition of resistance to PDT in response to TGFβ1. In addition, the sustained exposure to autocrine TGFβ1 signalling, as a result of the elevated endogenous production of this cytokine by SCC13 cells, may contribute to the reduced responsiveness to TGFβ1, in comparison to A431 cells. Nevertheless, there may be other important alterations involved. For this reason, future perspectives are focused on investigating the specific characteristics of different cSCC cell lines in relation to the TGFβ1 pathway and its mediators, as well as other players potentially involved in the regulation of the cell cycle progression and in the resistance to PDT.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







