1. Introduction
Canonical apoptosis occurs during regular tissue replacement as part of the normal physiological homeostasis. As apoptosis does not require any pathological warning signs to signal, it is a silent process. However, infections or cancer cell developments are pathological conditions. These settings must be recognized by immune surveillance, and this is the case, for example, in pyroptosis [
1]. Cell death processes rising in pathological conditions may signal danger signals recognized by specific immune cells to the immune system. Then the native or adaptive immune response is initiated against these pathological conditions leading to cell death. This cell death modality is called immunogenic cell death. The minimum requirement for immunogenic cell death is the plasma membrane permeabilization and releasing some intracellular damage-associated molecules (DAMs). ATP, calreticulin, the endoplasmic reticulum protein, HMGB1, the DNA binding nuclear protein, or the cellular RNA and DNA are typical DAMs [
2,
3,
4,
5]. Many regulated cell death processes share these primary conditions. For example, in pyroptosis, necroptosis, ferroptosis, or in lysosomal cell death, the plasma membrane gets permeabilized by specific membrane proteins or by oxidative or digestive degradation and opens the route for DAMs. Several harsh physical conditions also lead to the outer surface membrane rupture and the spillover of intracellular contents, such as the necrosis in the accidental cell death.
In the therapy of different cancers, such as skin, breast, or colon, the most established chemotherapeutic agents act in a DNA-damaging manner, leading to tumor cells’ apoptotic death [
2,
6]. However, activation of the typical apoptosis triggers a rather general anti-inflammatory immunological effect; therefore, killing cancer cells often go unnoticed by the immune system. Phototoxic agents are also effective DNA-damaging agents with the advantage that their action is controllable by light. On the other hand, phototoxicity can target cellular organelles besides DNA, thus acting not only through the destruction of rapidly dividing cells. Damage to extra-nuclear organelles usually causes pyroptosis or necroptosis, which are more immunogenic cell death mechanisms, triggering an adaptive immune response against the tumor cells [
3,
7,
8]. This type of phototoxicity provides an opportunity for the immune system to recognize cancer cells as foreign and eliminate tumors in an autonomous and specific manner. The significance of photodynamic therapy (PDT) comes from the fact that photosensitization causes inflammation, for example, in the skin and can activate the immune system [
9,
10,
11].
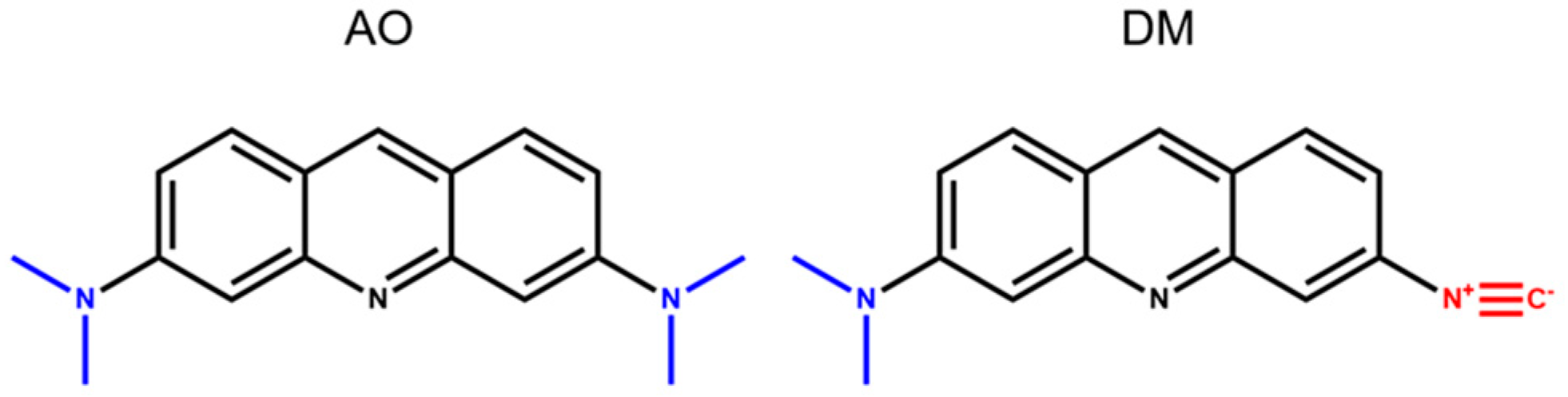
Acridine orange (AO,
Scheme 1) is a small molecular photosensitizer (PS) and can serve as a promising agent in PDT [
12]. The light-responsive properties and cellular activities of AO are relatively well understood. Although the PS activity of AO has been known for more than 100 years, its use in human PDT has only recently started to spread. The WHO classified AO as non-carcinogenic, and that helped the broadening of the clinical applicability of AO. In several short and long-term clinical trials, both topically and intravenously, AO is toxicologically safe in intact tissues. The medical applications of AO have been mainly in the field of theranostics [
13]. Due to its chemical nature, AO accumulates specifically in slightly acidic tumors, and according to its high fluorescence quantum efficiency, it can mark tumor tissues. AO has been successfully used for intraoperative identification of sarcomas or various other neoplasms having intestinal, cervical, or breast origin by precisely identifying tumor resection sites and enhancing tumor cell elimination using the illumination of the resection area during the operation.
AO stains the cytoplasmic and nuclear RNA of living cells green (orthochromatic) but does not bind to live cells’ nuclear DNA. Simultaneously, it colors the nuclear DNA of dying, apoptotic, or necrotizing cells green. AO changes its staining spectrum in fixed cells. In dying or fixed cells, AO binds to RNA as dimers (stacking two molecules), staining it intensive red (metachromatic). AO intercalates to the DNA of fixed cells as monomers and stains denatured DNA green [
16,
17]. Another interesting phenomenon suitable for studying lysosomal membrane integrity in live cells is the AO-sensitized photodestruction of lysosomes. In living cells with adequate metabolic activity, besides staining RNA green, AO accumulates in lysosomes. Concentrated AO in lysosomes also forms dimers, causing the characteristic metachromatic shift of the emission spectrum and stains lysosomal vesicles red. When cells are exposed to blue light, AO-generated radicals damage the lysosomal membrane; red vesicles turn to green firework-like flashes by exploding spectacularly as concentrated AO dilutes into the cytosol [
16,
18,
19,
20]. Proliferating tumor cells, according to the Warburg effect, are more acidic than normal tissue. Acidification explains the spontaneous accumulation of AO in neoplasms [
21]. Other tumor-related variations are increasing RNA and DNA content [
13] or multiplying intracellular membrane systems, such as the frequent expansion of lysosomes, all of which further strengthen the accumulation of AO in tumors. The overexpression of the vesicular autophagosome-lysosome system is particularly characteristic of tumors having multidrug resistance. In these tumors, chemotherapeutic agents primarily accumulate and are metabolized in lysosomes. A lysosomal exocytosis is a handy option for eliminating chemotherapeutic agents, representing an additional degree of freedom in tumor evolution. The AO can selectively recognize and attack the most resistant types of tumors through the PDT, making its research precious. To compare the action of AO with that of very closely related compounds would reveal even more in-depth insights into the cell biological mechanism of action of these photodynamic compounds and could help their use in clinics.
Recently, we reported the synthesis and characterization of a novel fluorescent dye family called amino-isocyanoacridines for pH sensing in the physiological range and live-cell imaging [
15]. These acridine derivatives showed altered properties compared to AO, such as reduced basicity, enhanced solvatochromic emission range, low quantum yield in water, and easy tuning of their fluorescent behavior by complexation with metal ions. It was also shown that among these novel derivatives, the 3-N,N-dimethylamino-6-isocyanoacridine (DM) has the most favorable properties in terms of excitation wavelength, fluorescent emission range, and preserving the original cell morphology. AO, compared to DM, being a symmetric molecule, can be considered almost nonpolar, especially in the ground state. In contrast, the substitution of one of the electron-donating (D) dimethylamino groups of AO (
Scheme 1) to an electron-withdrawing (A) isocyanide group enhances the polarity of the DM molecule considerably (the outwardly oriented, negatively charged end of the DM is the isocyano, while the positive is the dimethylamino group). The basis of the high polarity is the internal charge transfer (ICT) between the two different groups (D-A) through the aromatic acridine ring, which results in an enhanced dipole moment change (∆µ = µ
e − µ
g) between the excited and ground state (∆µ = 2 and ∆µ = 8 calculated in debye for AO and DM, respectively). The more considerable dipole moment change in DM is the reason for its enhanced solvatochromic range compared to that of AO and its potential application as pH probes for lysosomes at physiologic cellular conditions [
15]. Thus, we wondered whether DM could be an efficient lysosomal probe of human cells.
Acridine derivatives often bind to nucleic acids, thus frequently damaging the genetic material in PDT. This process, unfortunately, may lead to immunologically silent apoptosis. However, lysosomes serve as the extra-nuclear target of acridine derivatives, and their disruption directs the mechanism of cellular death towards the immunologically effective necrotic-like cell death. Therefore, it was interesting to compare the DNA-damaging and lysosome-destructive effects of AO quantitatively to that of our new DM compound.
To learn more about the mechanism of PDT action, we evaluated the cell biological properties of the AO and DM in detail. We ascertained that both AO and DM have comparable actions with slight alterations in some scenarios. AO affected the DNA more than DM, giving more extensive apoptosis-like DNA damage on the molecular level. The DM, however, caused more lysosomal injury and resulted in more massive and abrupt necrosis-directed cell damage. This behavior is rooted in their chemical structure; the DM has more significant polarity than AO, explaining its pronounced lysosomotropism and sharper imaging of lysosomes. Moreover, using this novel dye during the photodynamic cell treatment, the real-time release of damaged DNA from cell nuclei as shockwaves of DNA fragments could be visualized, as far as we know, for the first time.
2. Materials and Methods
2.1. Chemicals and Cells
3-N,N-dimethylamino-6-isocyanoacridine (DM) was obtained in a two-step reaction starting from 3,6-diaminoacridine. In the first step, one of the amino groups of 3,6-diaminoacridine was converted to isocyanide using dichlorocarbene formed in situ from chloroform in a basic medium. The product of the first step is 3-amino-6-isocyanoacridine (ICAAc). During the second step, the methylation of the free amino group of ICAAc was carried out with CH3I/KOH in toluene. The pure dimethylamino derivative (DM) was obtained after purification by column chromatography. The detailed synthetic procedures and characterization of DM and the intermediates are found in the Supporting Information of [
15]. All chemicals were from (Sigma Aldrich, Budapest, Hungary) unless otherwise stated.
The HeLa (#CRM-CCL-2, ATCC) human cervix epithelial adenocarcinoma cell line and the SK-BR-3 (#HTB-30, ATCC) human breast cancer cell line, here labeled as SKBR-3, was obtained from the American Type Culture Collection (Rockville, MD, USA). The OCM-1 human melanoma cell line was kindly provided by Dr. H. M. H. Hurks, Department of Ophthalmology, Leiden University Medical Center, Leiden, The Netherlands [
22]. The 3T3-MDR1 cell line is the human ABCB1 protein transfected NIH 3T3 mouse fibroblast cell line expressing a high level of the human transporter (named originally as NIH 3T3 MDR1 G185 cells) is a kind gift from Michael Gottesman (National Institutes of Health, Bethesda, MD [
23]. OCM-1 cells were maintained in RPMI, while other cell lines were maintained in DMEM (Livetech, Raleigh, NC, USA). Both media were supplemented with 10% fetal calf serum (Thermo Fischer Scientific, Budapest, Hungary), 2 mM glutamine, PS (10 U/mL penicillin, and 10 µg/mL streptomycin) or 10 µg/mL gentamycin, and with Phenol-Red (Livetech) in a humidified atmosphere with 5% CO
2 at 37 °C.
2.2. Cellular Toxicity Measurements
For cellular toxicity measurements, 10,000 cells were seeded in each well in a Corning Costar 96 well plate. Dilution series of 9.4 × 10
−5, 3.8 × 10
−5, 1.9 × 10
−5, 9.4 × 10
−6, 4.7 × 10
−6, 2.4 × 10
−6, 1.2 × 10
−6, 5.9 × 10
−7, 2.9 × 10
−7, 1.5 × 10
−7 M from AO and 1.0 × 10
−4, 4.0 × 10
−5, 2.0 × 10
−5, 1.0 × 10
−5, 5.1 × 10
−6, 2.5 × 10
−6, 1.3 × 10
−6, 6.3 × 10
−7, 3.2 × 10
−7, 1.6 × 10
−7 M from DM were applied in four parallel experiments. After the 24 h incubation time, 100 µL MTT solution (0.5 mg/mL in PBS: 150 mM NaCl, 3.3 mM KCl, 8.6 mM Na
2HPO
4, and 1.69 mM K
2HPO
4, pH 7.4) was added to each well. The plates were incubated for 2 h at 37 °C, then the wells were aspirated, and the MTT formazan was extracted with 200 µL of DMSO aided by gentle agitation on a shaker. After 30 min at room temperature, a Synergy HT multi-detection microplate reader (BioTek, Winooski, VT, USA) recorded the absorbance values at 565 and 680 nm. The instrument was blanked beforehand on a row not containing cells, and the absorbance was corrected by subtracting absorbance values measured at 680 nm from the corresponding absorbance of 565 nm. Percentage viability (respiratory competence) of the population of cells in each well was expressed as:
2.3. Spectrofluorimetry Measurements
The excitation and emission spectra were recorded in a quartz cuvette of 1.0 cm optical length using a Jasco FP-8200 fluorescence spectrophotometer (JASCO Corporation, Tokyo, Japan), equipped with a Xe lamp light source. Data were recorded at 20 °C, using 2.5 nm excitation, 5.0 nm emission bandwidths, and 200 nm/min scanning speed at “normal” sensitivity. First, we recorded all spectra of solutions containing one million HeLa cells/mL labeled with 2.4 × 10−6 M AO or 2.5 × 10−6 M DM. Then, in control experiments, these parameters were recorded with the same settings but without the cells.
2.4. Confocal Microscopy and Spectral Imaging
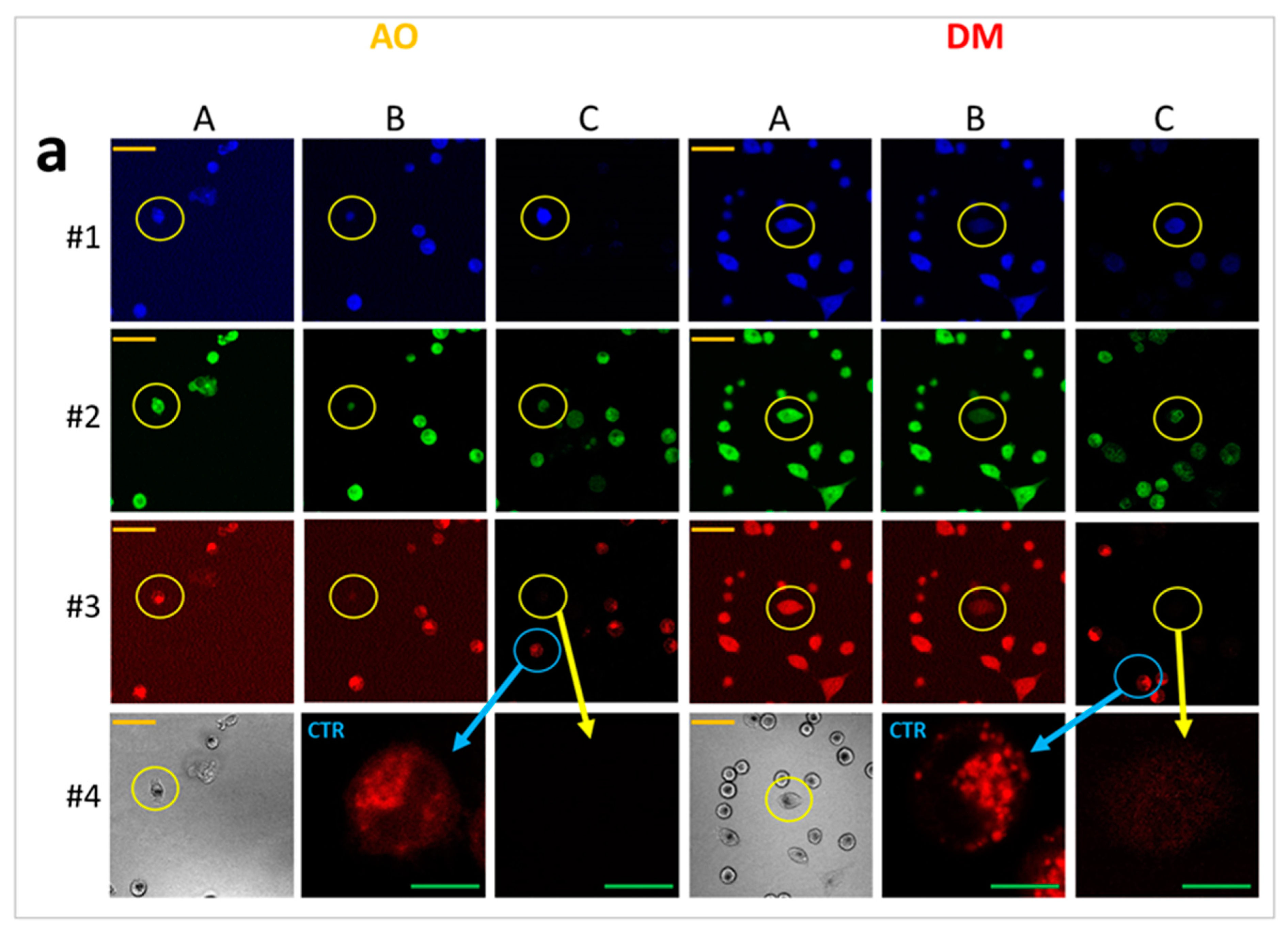
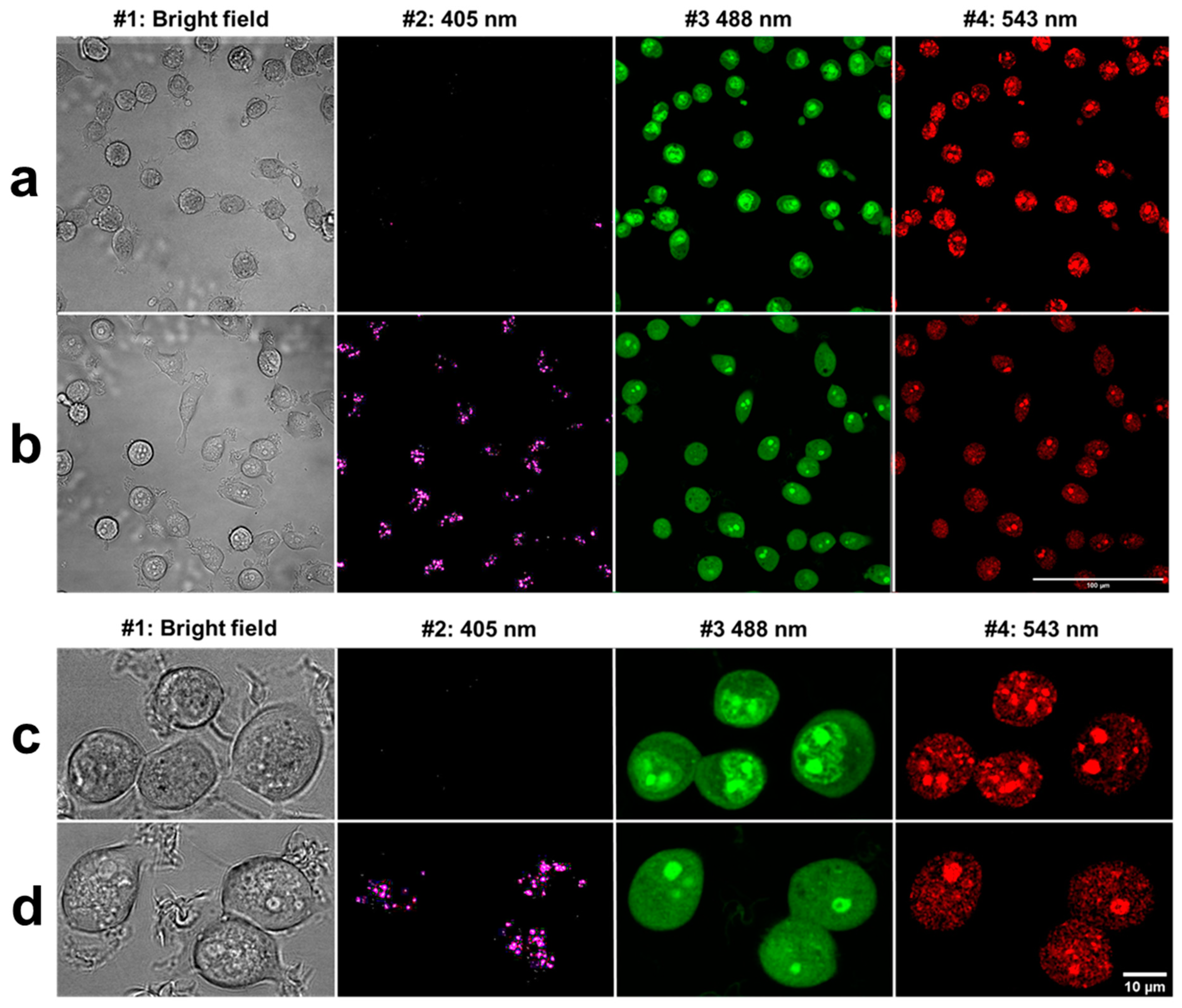
Cells were seeded in ibidi slide (8-well ibidi µ-Slide, Zenon Bio Kft, Szeged, Hungary) at a concentration of 30,000 cells/well and the next day labeled with 2.5 × 10−6 M AO and DM for 2 h. Then, fluorescence and bright field 16-bit images were taken by a Zeiss LSM 880 confocal microscope (Carl Zeiss microscopy Gmbh, Oberkochen, Germany) using a ×40 C-Apochromat water immersion objective applying 405, 488, and 543 nm laser excitation and blue (410–474 nm), green (499–540 nm), and red (554–629 nm) emission channels. For spectral imaging, the same wavelengths excitation laser lines were applied, and total emission ranges at the three excitations were 410–695, 499–695, and 553–695 nm, respectively. A fair number of images were made at every 18-nm span with a 32-channel linear detector system in these broader ranges.
2.5. Investigation of Lysosomal Morphology Applying LysoOrange
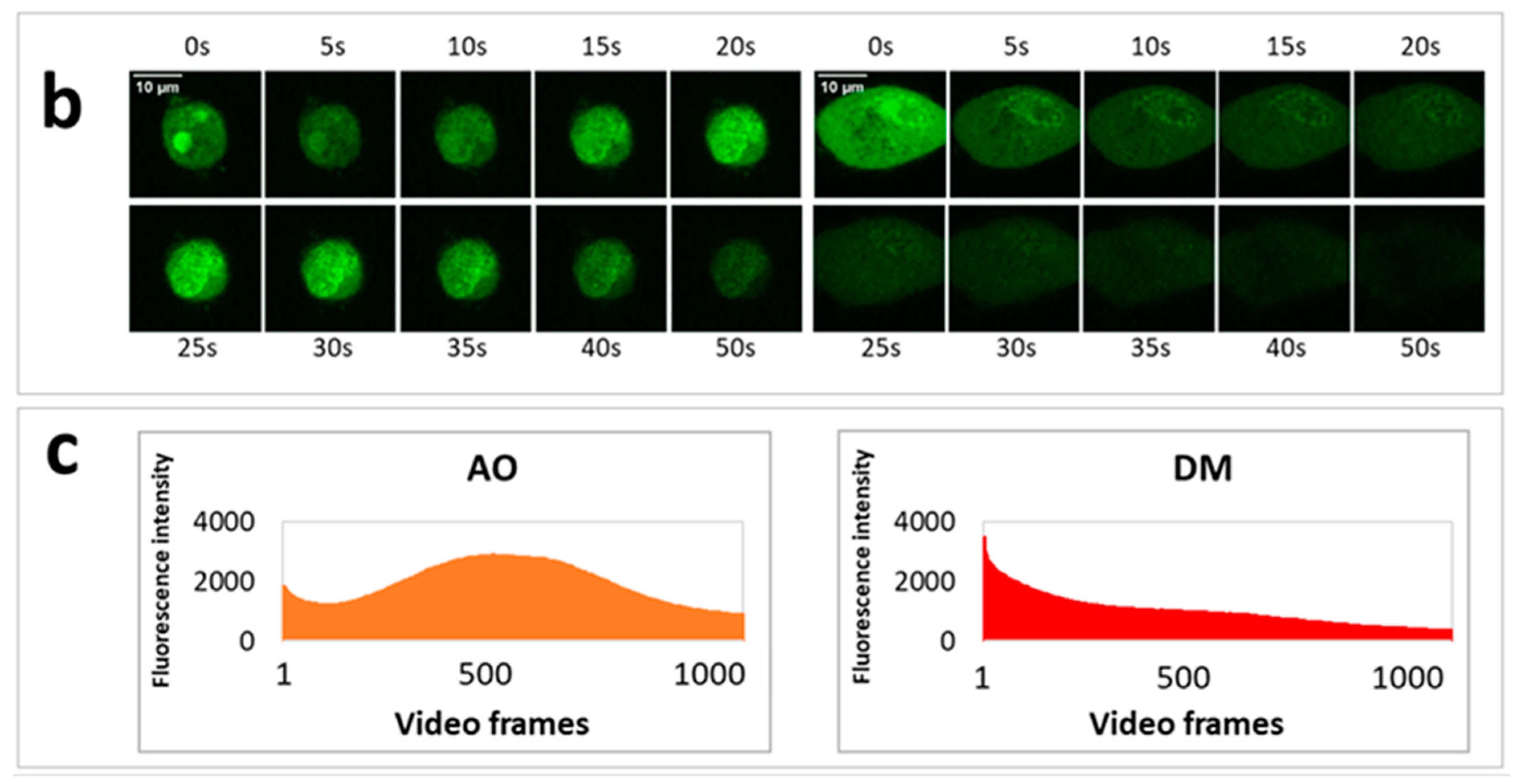
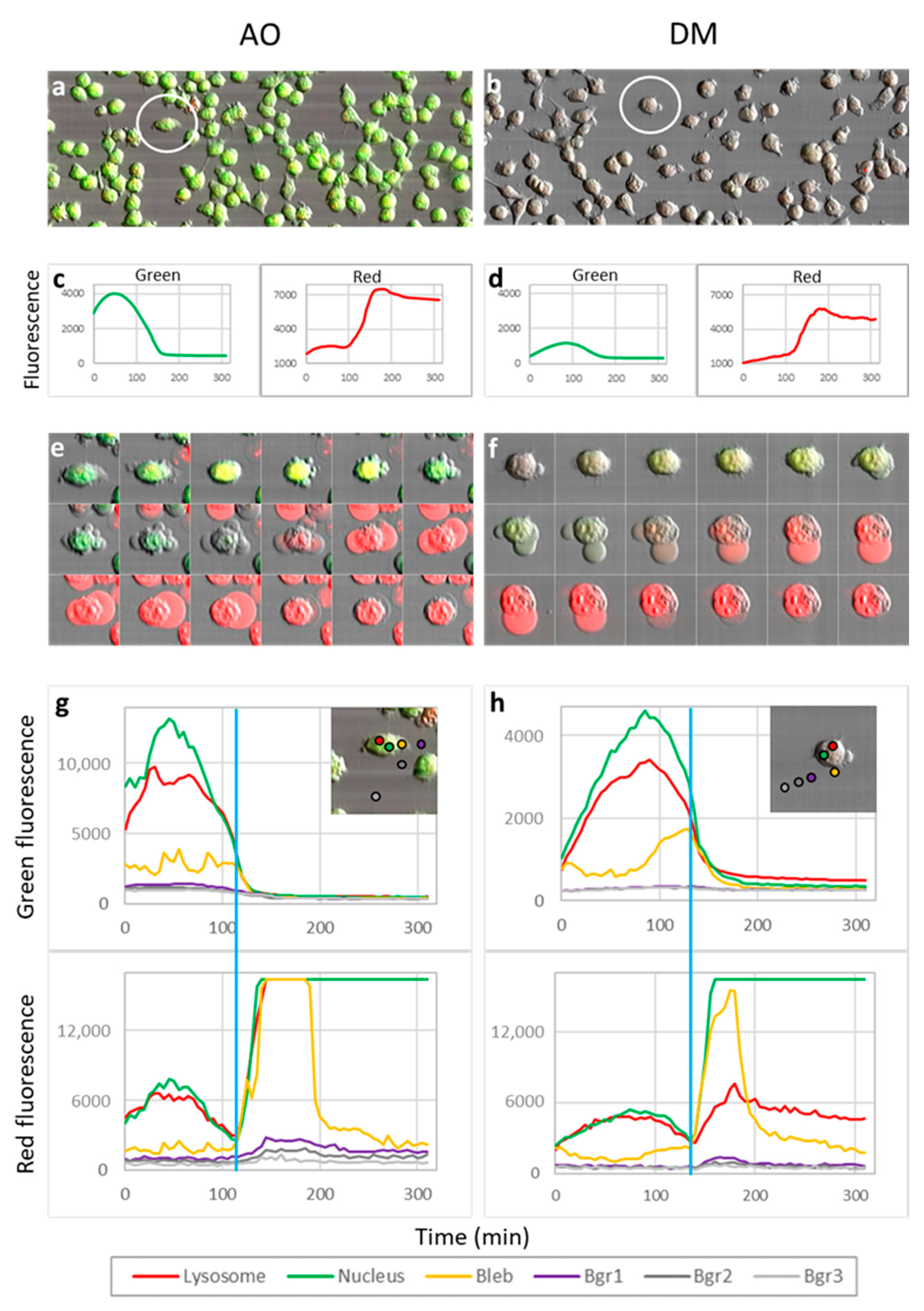
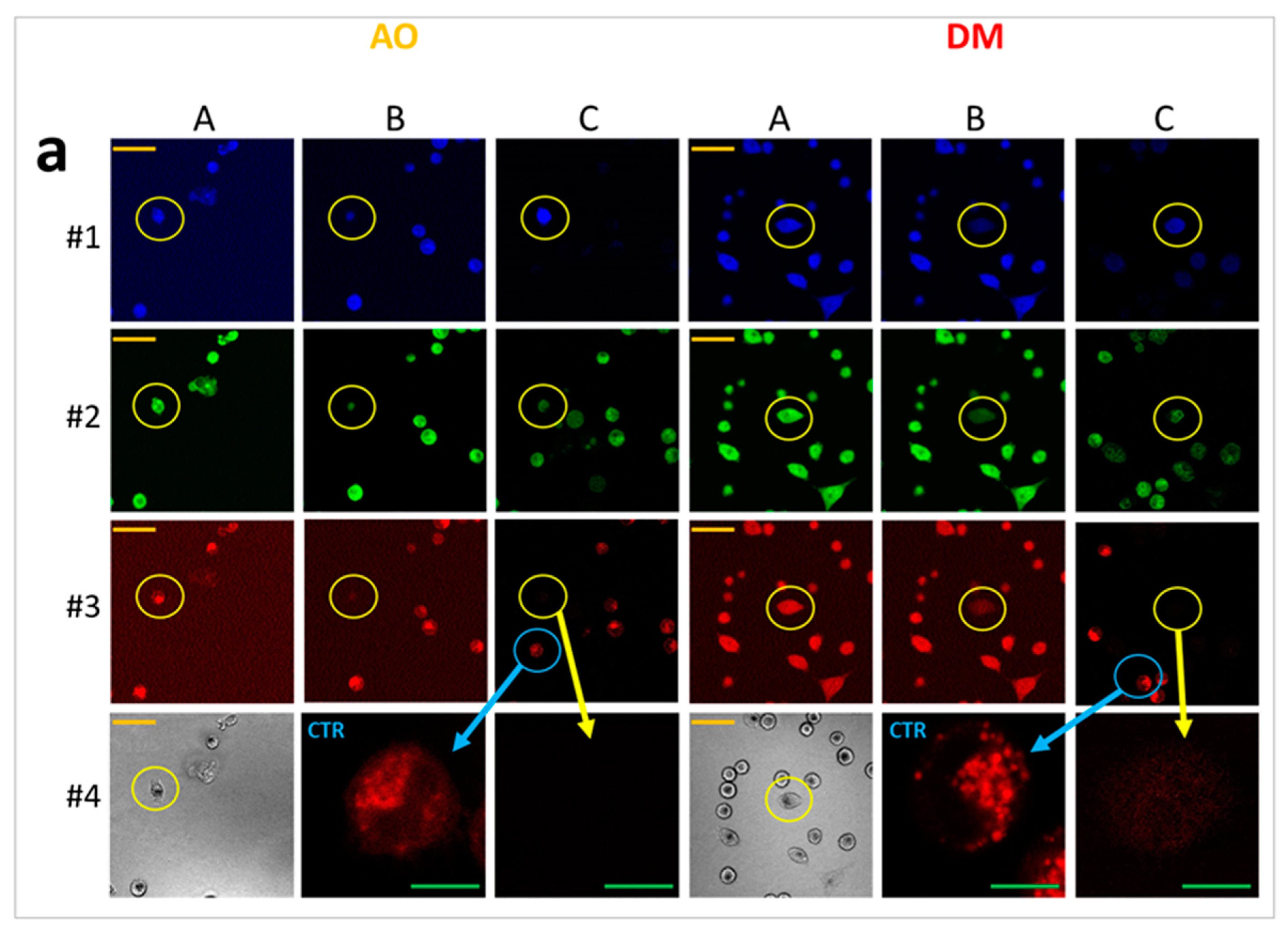
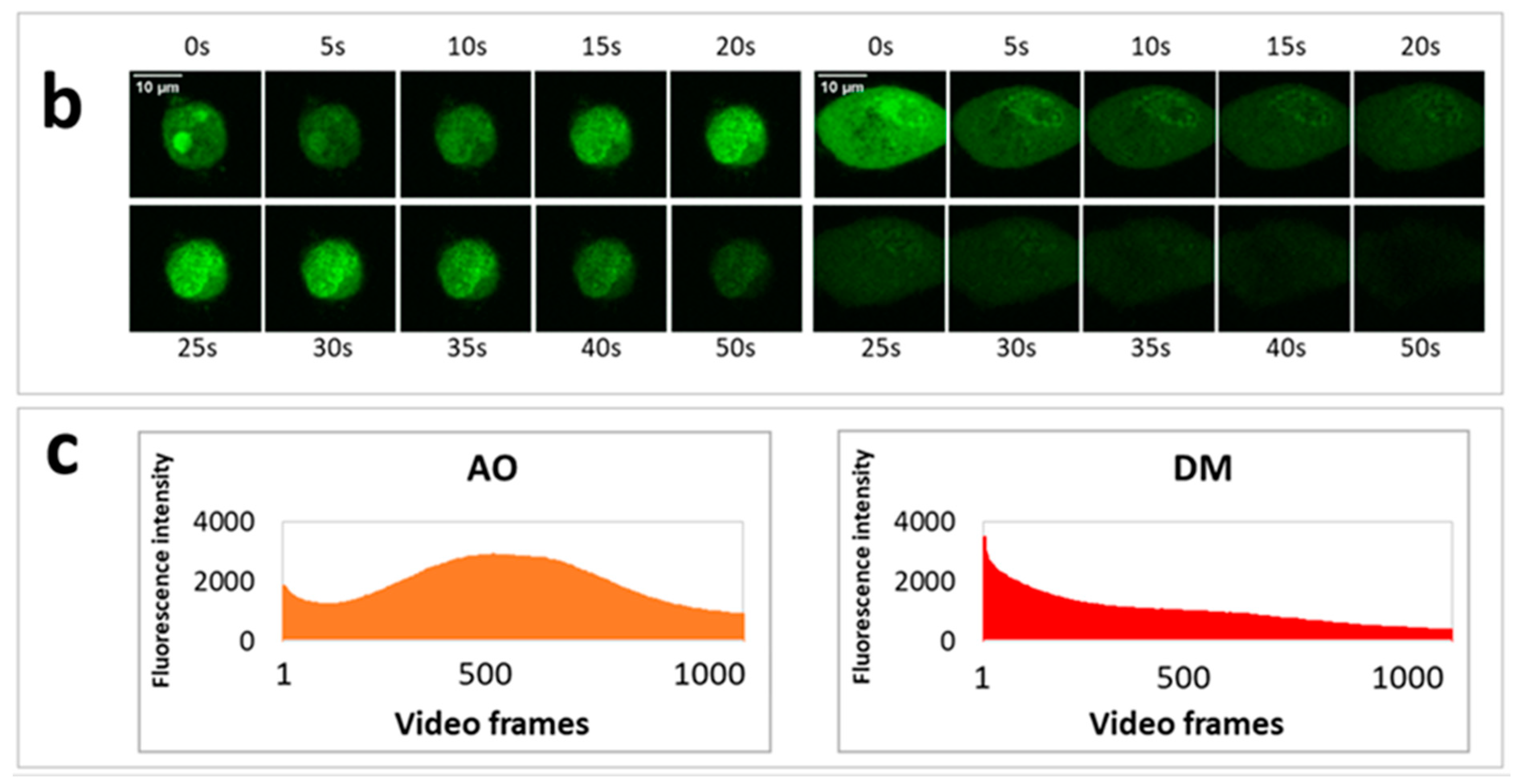
On the previous day of the measurement, 30,000 HeLa cells were seeded to each well of an eight-well ibidi slide. In order to visualize lysosomes, we stained the cells with a sub-vital lysosomal probe in a humidified atmosphere with 5% CO2 at 37 °C. 20 µL LysoOrange (ab176827 CytoPainter, Abcam, Cambridge, UK) was added to 10 mL RPMI medium, and from this lysosome labeling solution, 300 µL was used to an ibidi well. After 15 min of incubation, images of cells were recorded at 543 nm excitation wavelength. Testing the effect of PS and light on lysosomal morphology, one plate was treated with 2.5 × 10−6 M AO and another with the same concentration of DM. The incubation time was one hour under the same cell culture conditions. After the incubation with PS, one cell was selected, and an image was taken (before irradiation). Then, with 50-s continuous imaging, 1000 frames were recorded (illumination), and a 20 frame per second (fps) video was constructed from these images of the same cell. After the illumination, the medium with the PS was replaced by the lysosome labeling solution. After the lysosomal staining at 37 °C, another image was taken from the same cell. The procedure was repeated both for the AO and DM.
2.6. Phototoxicity Measurements with Blue LED Lamp Applying MTT Bioassay
Cells were seeded at 10,000 cell/well concentrations one day before the experiment. To test the phototoxicity of the compounds, we set up the following system. A 470 nm wavelength LED lamp (Alustar 900808; Ledxon, Geisenhausen, Germany) was placed 10 cm above and oriented towards the sample in a 96-well plate situated in a conventional cell culture incubator. The optical axis of the illumination was centered to the center of the flat-bottom plate. For the light treatment, the attached cells in different plates were irradiated for increasing times. Cell viability was determined by applying the MTT assay, as described above. The blue LED lamp had a 30-degree cone with a 23.3 lm luminous flux, which, according to our calculations, corresponded to a 4.45 mW/cm2 fluence rate at the sample level.
2.7. Laser-Scanning Cytometry (LSC) and Live-Cell Imaging
Laser-scanning cytometry was used for fluorescent live-cell imaging with parallel fluorescent detection of cell death events and video recording; QUINESIn measurements were utilized to evaluate DNA superhelicity, histone quantities, and DNA integrity in single cells [
18]. The slide-based laser-scanning iCys Research Imaging system (Thorlabs Imaging Systems, Sterling, VA, USA) was equipped with four lasers, two photodiodes for chromatic absorbance detection, and four photomultipliers for fluorescence detection, and with an ibidi microscope-stage incubator (Zenon Bio Kft, Szeged, Hungary). Imaging of the samples was set up in one or several predefined regions of interest (ROI), where scanning took place point by point, either at once or separately following one another by the multiple laser lines. A fixed offset from the bottom of the well was applied during scans focusing on the middle plane of cells. User-defined areas in the specimen with optimal cell density were marked as regions of interest and scanned in an automated manner.
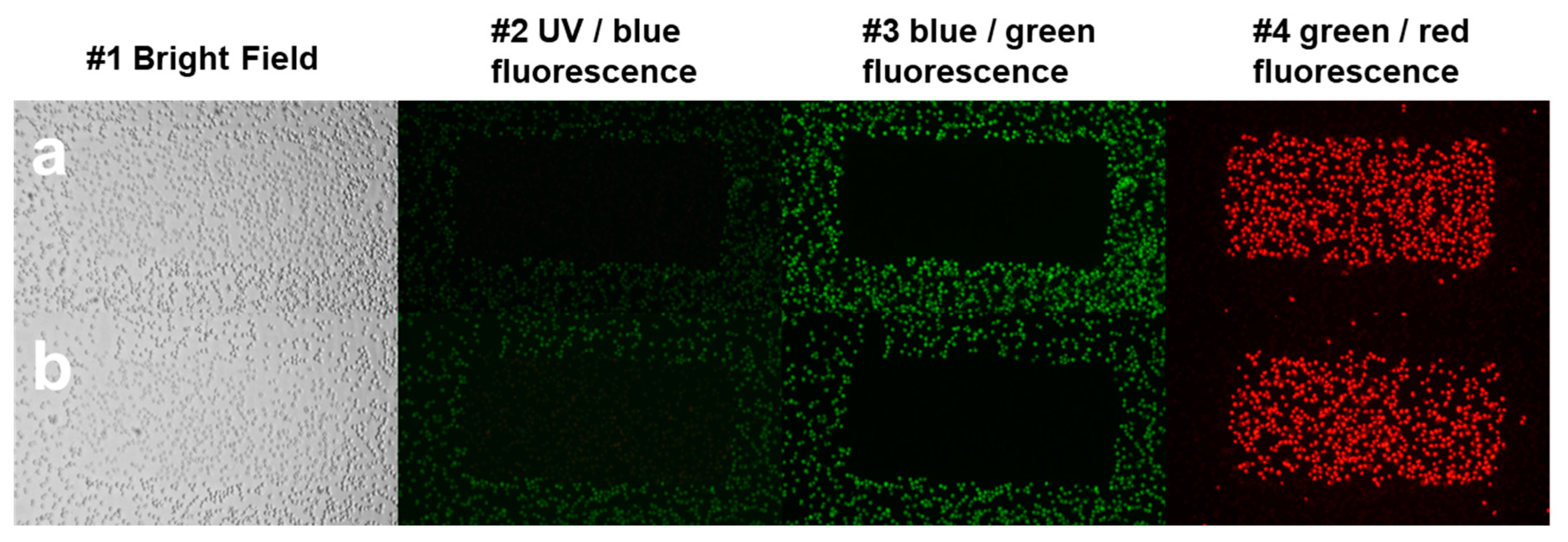
For live-cell imaging and video recording, cells were kept at 37 °C with 5% CO2 air condition and 90% humidity. Cells were seeded at 30.000 cells/well into ibidi slides 24 h before the measurement, and cells were treated with 2.5 × 10−6 M AO or DM. Propidium iodide was added to each sample at 7.5 × 10−6 M final concentration. One field image was recorded at the region of interest (ROI), with a 500 × 184 μm2 area. The field image was 1000 × 768 pixels; hence the size of one pixel was 0.5 × 0.24 μm. One laser-scanning illumination cycle lasted for 2.9 s, and illuminations were replicated every 5 min. Imaging was repeated in 63 consecutive cycles. The arising fluorescence signals of the 405, 488, 561, and 633 nm lasers were collected by a 40∙LWD (NA 0.6) objective into four detection channels (wavelengths are given as mean/range in nm): blue (480/40 nm), green (530/30 nm), red (580/30 nm), and long-red (675/50 nm). In some live-cell experiments, just the 488 nm laser and the green and far-red channels were used. The measured laser power of the 488-laser line was 0.30 mW ± 0.05 (mean ± SD), which provided a 0.32 W/cm2 fluence rate at the sample. After the LSC scan, wide-field fluorescence images were taken by an Olympus DP71 color camera (Olympus Hungary Kft., Budapest, Hungary) applying typical violet/blue, blue/green, and green/red filter cubes with mercury vapor lamp illumination.
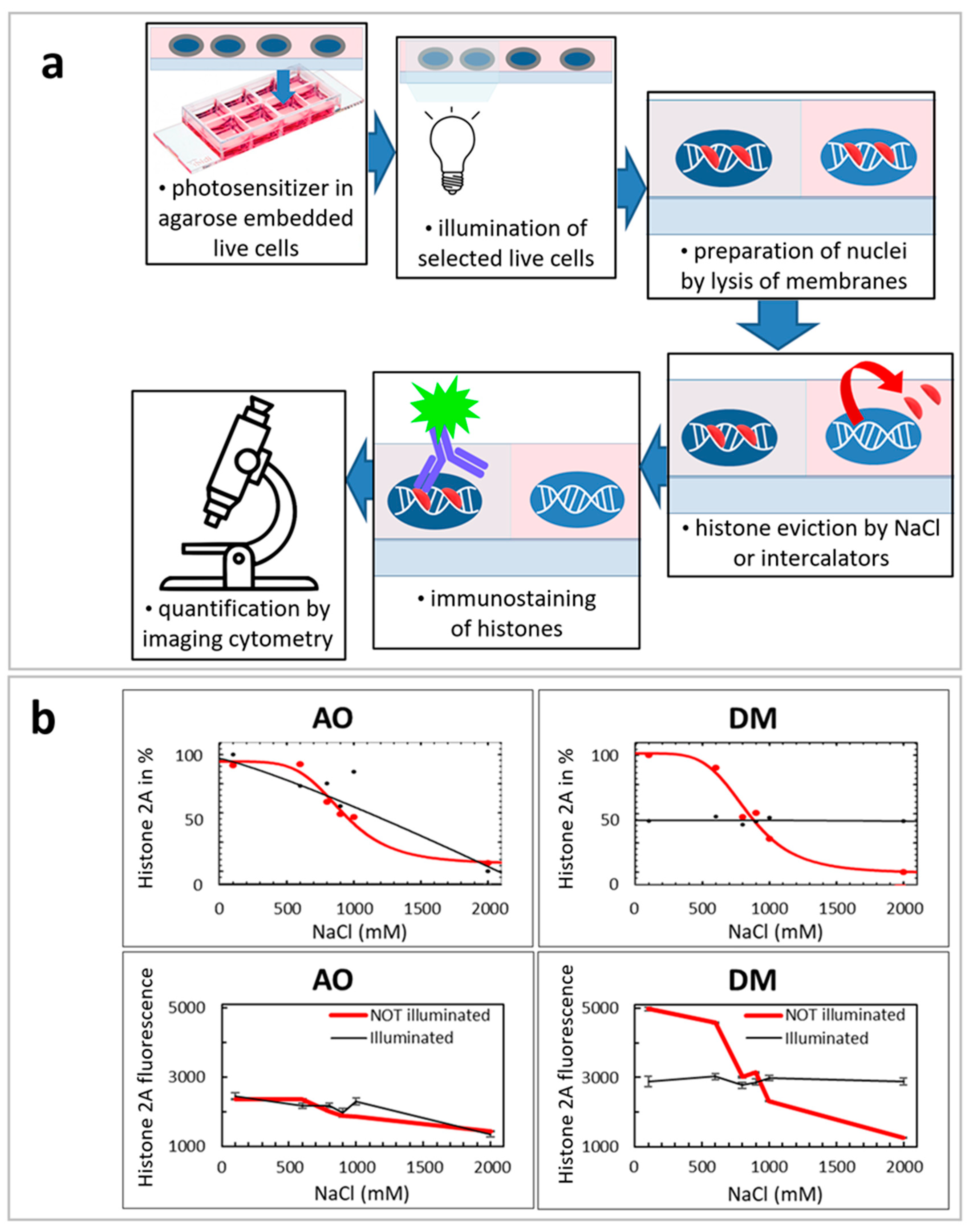
2.8. Illuminated QUINESin Method
We described earlier the original QUINESin method [
24]. In this work, we slightly modified this method to verify the effect of light illumination for histone elution (Figure 6a).
In the beginning, live cells were embedded into low melting point agarose. Before embedding, the wells of ibidi slides were coated with 1% (m/v) low melting point agarose. A total of 150 μL liquid agarose, diluted in distilled water, was dispensed into each well and was immediately removed so that a thin agarose layer remained on the surfaces and was left to solidify on ice for 2 min. Then, slides were kept at 37 °C until the surface of the wells dried out. This coating procedure was repeated once more on the same chambers. Embedding was performed, keeping cells and agarose at 37 °C. The cell suspension containing 6 × 106 cells/mL was mixed with 1% low melting point agarose diluted in PBS at a v/v ratio of 1:3. 22 μL of the cell agarose suspension was dispensed in the middle of the wells and the chambers were covered with homemade rectangular plastic coverslips cut out from a 200 μm thick, medium weight polyvinyl chloride binding cover (Fellowes, Inc., Itasca, IL, USA). The cells were left to sediment on the coated surface of wells for 4 min at 37 °C and then kept on ice for 2 min. When the agarose solidified, a 300 μL ice-cold complete culture medium was added to each well, a step aiding the removal of coverslips.
2.8.1. Illumination of Agarose Embedded Live Cells with LSC for the Illuminated QUINESin Assay
After live-cell embedding, an area of one field image was irradiated by the 488 nm laser light in each well by making images with the LSC system (Figure 6a). The illumination of the exact location and the imaging was repeated in subsequent consecutive cycles, as described above in the LSC section.
2.8.2. Preparation of Nuclei/Permeabilization and Histone Eviction by Salt or Intercalators
The agarose-embedded cells at the bottom of the wells were washed with 500 μL ice-cold PBS three times for three minutes, then permeabilized with 500 μL ice-cold 1% (v/v) Triton X-100 dissolved in PBS/EDTA (5 mM EDTA in PBS) for 10 min. This step was repeated once more. After permeabilization, the nuclei were washed by a regular three-minute in-well bath with 500 μL ice-cold PBS/EDTA three times and were treated with different NaCl solution concentrations on ice. The nuclei were washed with 500 μL of ice-cold salt solution for 60 min. After this step, the nuclei were bathed in-well with 500 μL of ice-cold PBS/EDTA three times. NaCl was diluted in PBS/EDTA; the salt concentrations indicated on the X-axes of the graphs in all panels of Figure 6b show the total NaCl concentrations together with NaCl present in the PBS buffer. Analyses of the curves were performed by SigmaPlot 12.0, using either ‘Sigmoid 3 parameter’ (in the case of linear plots) or ‘Standard curves: Four Parameter Logistic Curve’ (in the case of logarithmic plots) curve-fitting subroutines. The number of analyzed G1 nuclei was between 200 and 1000 cells/well, out of the about 500–2000 cells scanned. All SEM values in Figure 6b were calculated from the cell population’s data points analyzed in the given experiment.
2.8.3. Immunofluorescence Labeling
After salt treatment, the samples were incubated with 500 μL 5% (m/v) Blotto Non-Fat Dry Milk (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) PBS/EDTA for 30 min on ice to decrease nonspecific binding of the antibodies. The blocking solution was washed out by an in-well bath with 500 μL ice-cold PBS/EDTA three times. Indirect immunofluorescence labeling was performed using rabbit polyclonal anti-H2A (Abcam) primary antibody, diluted in 150 μL of PBS/EDTA/1% BSA (PBS/EDTA supplemented with 1% w/v bovine serum albumin), at 4 °C, overnight at a titer of 1:800. After labeling with the primary antibody, the nuclei were washed with 500 μL ice-cold PBS/EDTA three times for 10 min. Labeling with the secondary antibody was performed in 150 μL PBS/EDTA for two hours on the ice, using Alexa fluor 647 conjugated goat anti-mouse IgG or goat anti-rabbit IgG antibodies (Thermo Fisher Scientific, Budapest, Hungary). The secondary antibody was also used at a titer of 1:800, diluted in PBS/EDTA from 2 mg/mL stock solutions. After labeling with the secondary antibody, the agarose-embedded nuclei were washed with 500 μL ice-cold PBS/EDTA three times for 10 min. The samples were then fixed in 1% formaldehyde (dissolved in PBS/EDTA) at 4 °C, overnight. After fixation, the embedded nuclei were in-well washed with 500 μL ice-cold PBS/EDTA three times and stained with 200 μL 5 μg/mL propidium iodide (dissolved in PBS/EDTA) for 30 min on the ice. The stained nuclei were incubated in the well with 500 μL ice-cold PBS/EDTA three times. Fluorescence intensity distributions were recorded using the LSC imaging system, as described above.
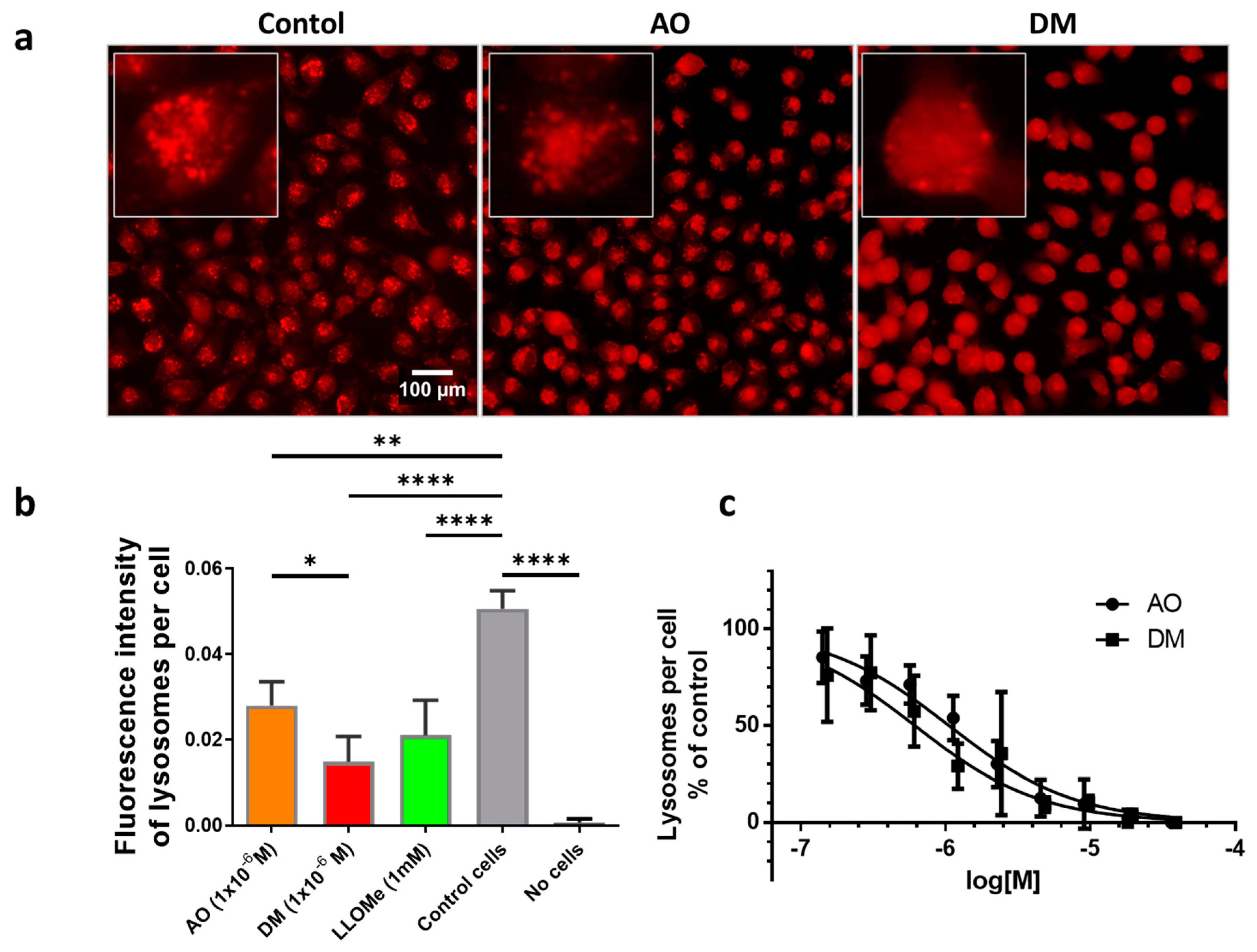
2.9. Quantification of Lysosomes and Lysosomal Membrane Integrity
One day before the measurements, 10,000 HeLa cells were seeded to each well of 96-well plates. The dilution series of 3.8 × 10−5, 1.9 × 10−5, 9.4 × 10−6, 4.7 × 10−6, 2.4 × 10−6, 1.2 × 10−6, 5.9 × 10−7, 2.9 × 10−7, 1.5 × 10−7 M from AO and 4.0 × 10−5, 2.0 × 10−5, 1.0 × 10−5, 5.1 × 10−6, 2.5 × 10−6, 1.3 × 10−6, 6.3 × 10−7, 3.2 × 10−7, 1.6 × 10−7 M from DM was applied in four parallel plates. As an untreated control, glucose-PBS (PBS supplemented with 8 mM glucose) was added to cells; the blank contained only glucose-PBS without cells. As for positive controls, cells were treated with LLoMe (L-leucyl-L-leucine methyl ester, Sigma Aldrich, in 25 mM), a known lysosomal damaging agent. After 45 min of incubation in a humidified atmosphere with 5% CO2 at 37 °C, the fluorescence intensity of the wells was measured by the Synergy HT multi-detection microplate reader. The excitation filter was 485/20 in both cases, while the emission filter was 528/20 for AO and 590/20 for DM. The fluorescence intensity parameters in the presence of photosensitizers were measured 10 times in each well in order to damage lysosomal membranes. Then solutions were removed, and the cells were stained with LysoOrange suspension similar to confocal microscopy. Wide-field fluorescence micrographs were finally taken from each well with the LSC imaging system color camera using green/red filter cubes and 20× objective described above.
2.10. Image Analysis of Cellular Lysosome Content
We have determined the lysosome content of cells of camera images for every well. The lysosome content of cells is determined in the images of wells as a quotient of the sum of pixels in every lysosomal region and all cells’ pixels. This method gives an approximation for the lysosome-cell ratio in an image. A detailed description of the image analysis is provided in the
supplementary materials.
2.11. Movies
Images were captured at defined intervals by imaging cytometry or confocal microscopy, then processed with the software of the systems and used to make original video frames. After further image enhancements, we have produced final movies played at a rate of 7 frames per second (fps) using the Fiji program.
2.12. Statistical Calculations
For data comparison, we have applied the Student’s t-test, Tukey’s multiple comparisons test, nonlinear regression dose-response inhibition semi-logarithmic normalized curve fitting, and comparison of fits statistical analysis using the GraphPad Prism 8.4.3 for Windows software.
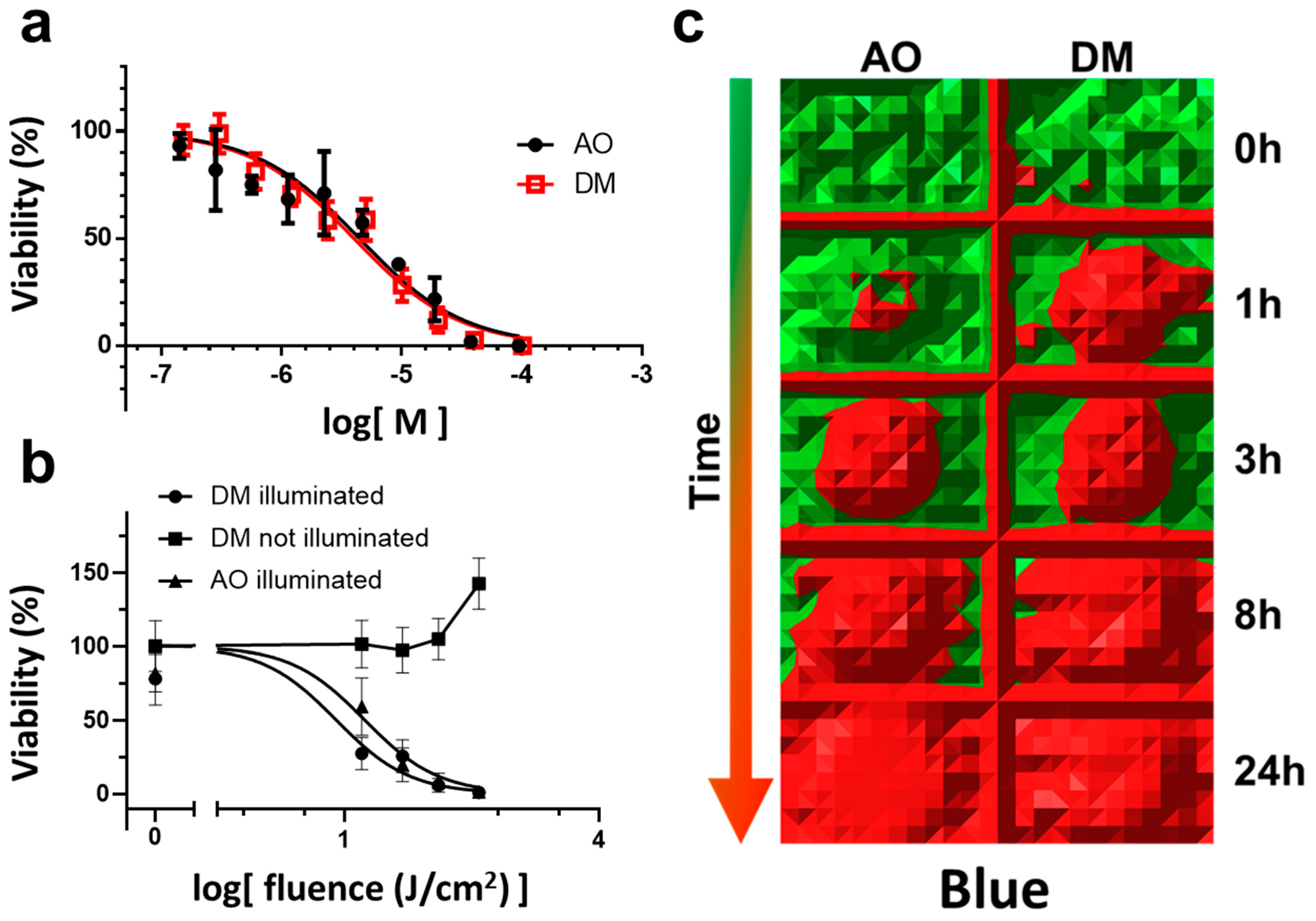
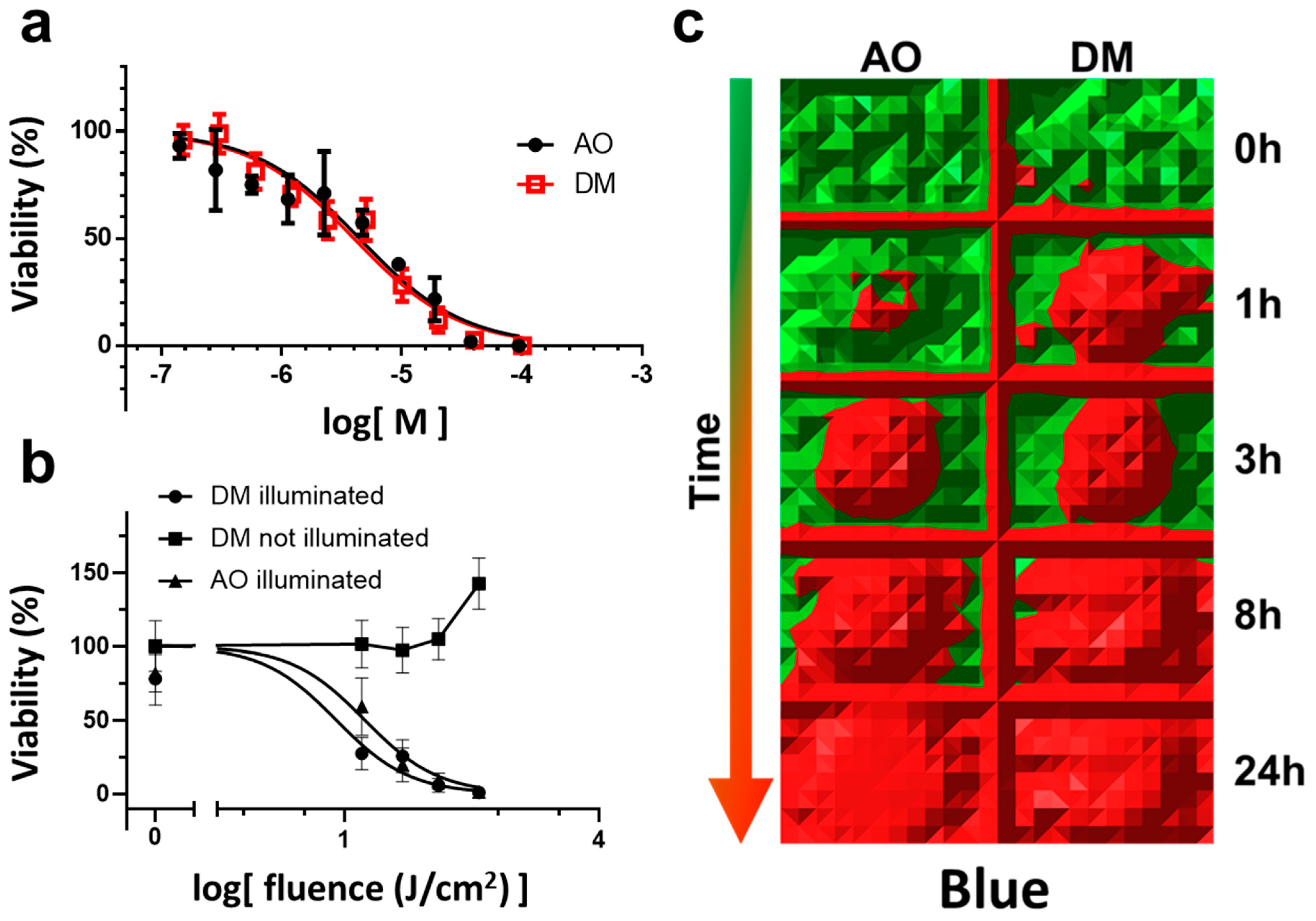
In detail, for the AO and DM toxicity measurements in the dark, four experiments were conducted (
Section 3.2, Figure 1a). A representative with four parallels for the selected drug concentrations was evaluated in GraphPad. The nonlinear regression dose-response inhibition semi-logarithmic normalized curve fitting was performed. The statistical evaluation of the comparison of fits did not show a significant difference.
For continuous LED light phototoxicity experiments, a representative of three experiments was selected and presented in Figure 1b,c (
Section 3.3). Here an entire 96-well plate with 2.5 µM drug treatment (AO and DM) was used for one time-point of the phototoxic kinetic measurement (Figure 1c) applying the MTT viability assay. At one time-point, the MTT values of the middle 36 wells (the center of all 96-well plates was centered to the optical axis of the LED) of the entire plate were first normalized to the average MTT value of one full unilluminated plate of 96 wells. Then the normalized 36 values were averaged. The mean and SEM of these normalized data were plotted (y viability value) as one fluence value in Figure 1b. The irradiation value (x fluence value of Figure 1b) was calculated as a 10-base logarithm of the product of the time of illumination in seconds and the fluence rate (mW/cm
2) of the illumination (calculation details in
Section 2.6). Then, DM and AO normalized curves were calculated and fitted according to the log(dose)-response inhibition model, and IC50 values were statistically compared using the F test for sum-of-squares in GraphPad. The
p-value was less than 0.0001.
Five experiments were conducted for LSC phototoxic experiments (
Section 3.6,
Section 3.7 and
Section 3.8) and two videos constructed with Fiji. One video was selected for quantification. All image fields contained approximately 70–150 dye-loaded cells. Dye content representing intensity profiles was measured on whole images (Figure 5c,d) or in the selected ten-pixel diameter circular region of interest (Figure 5g,h).
For QUINESIn experiments (
Section 3.10), we have selected one representative example out of the three similar experiments for Figure 6. On plots of Figure 6b, every data point was calculated from 50–500 individual cell nuclei.
For lysosomal photodamage experiments, plate-reader experiments were repeated three times. One plate was chosen, and images were recorded for all of the 96-wells. These images were processed for further image analyses, as detailed in the
supplementary materials. For statistical analysis of data in Figure 8b, AO and DM samples were compared by
t-test. Tukey’s multiple comparisons test was performed for all the samples. GraphPad’s curve fitting was performed by nonlinear regression using the log(inhibitor) dose vs. normalized response model. The fitted curves least-square data were tested using the F test (Figure 8c).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}