
Kinase Inhibitors’ Effects on Innate Immunity in Solid Cancers

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Innate Immunity in the Context of Cancer
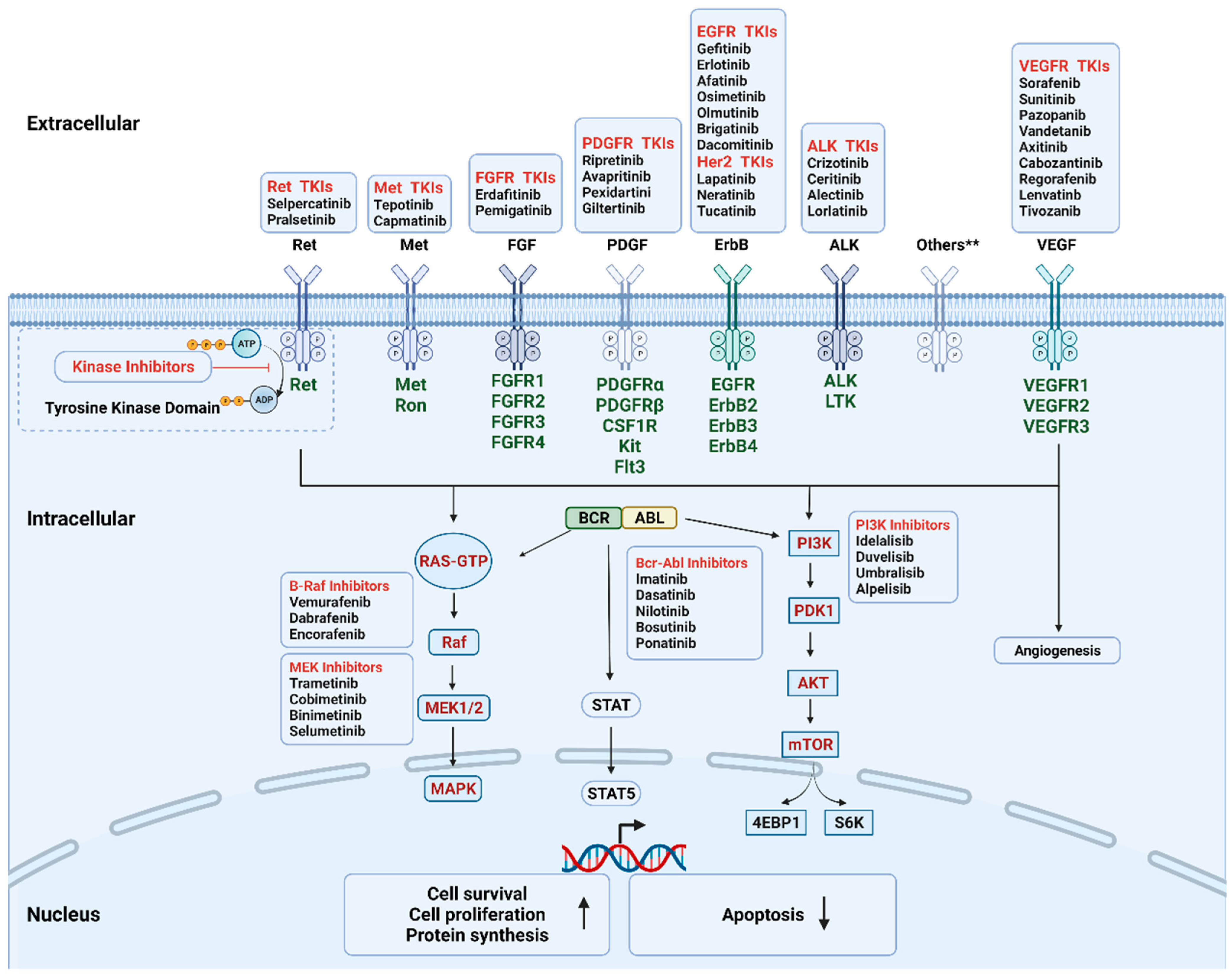
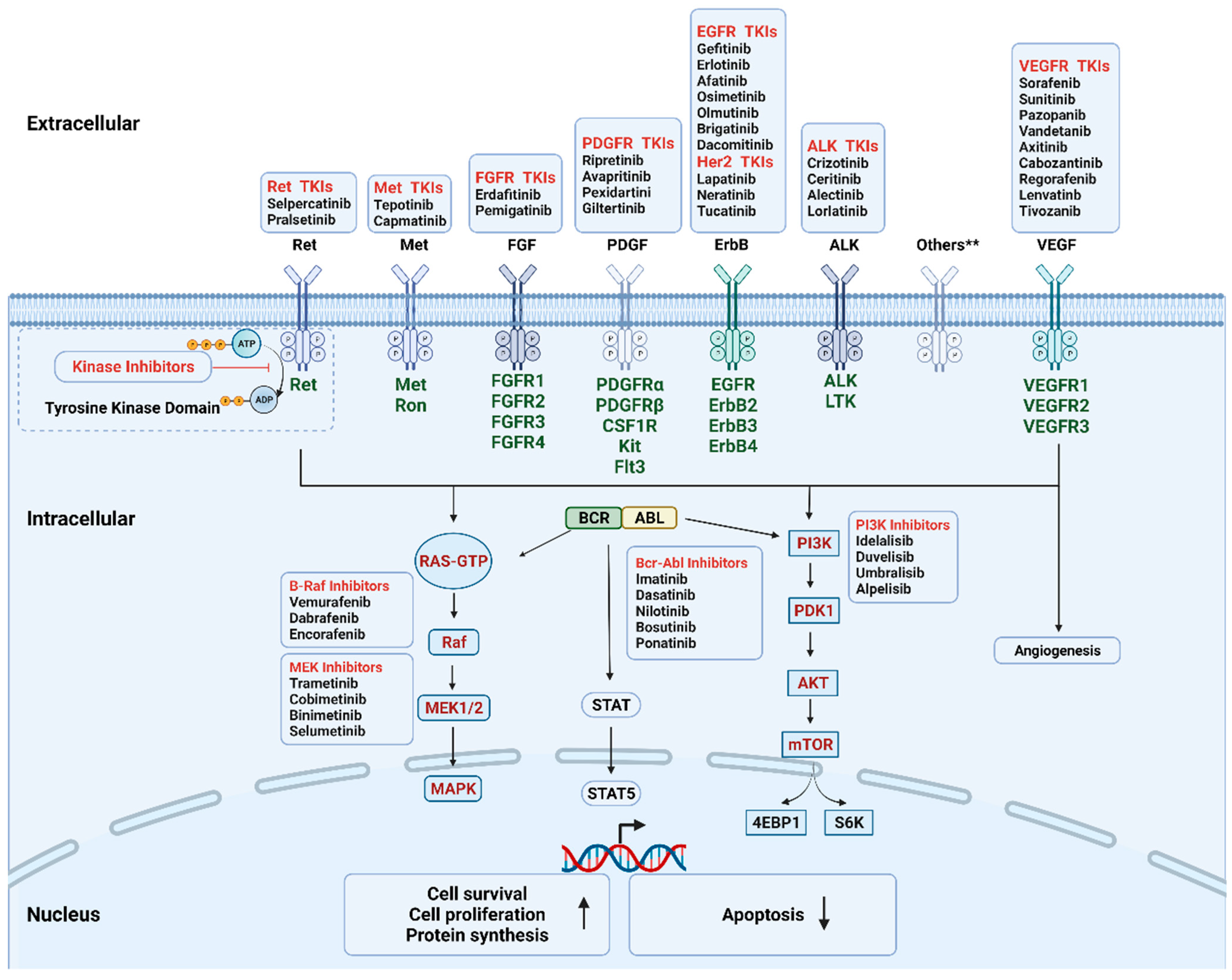
3. Kinase Inhibitors (KIs) for Cancer Treatment
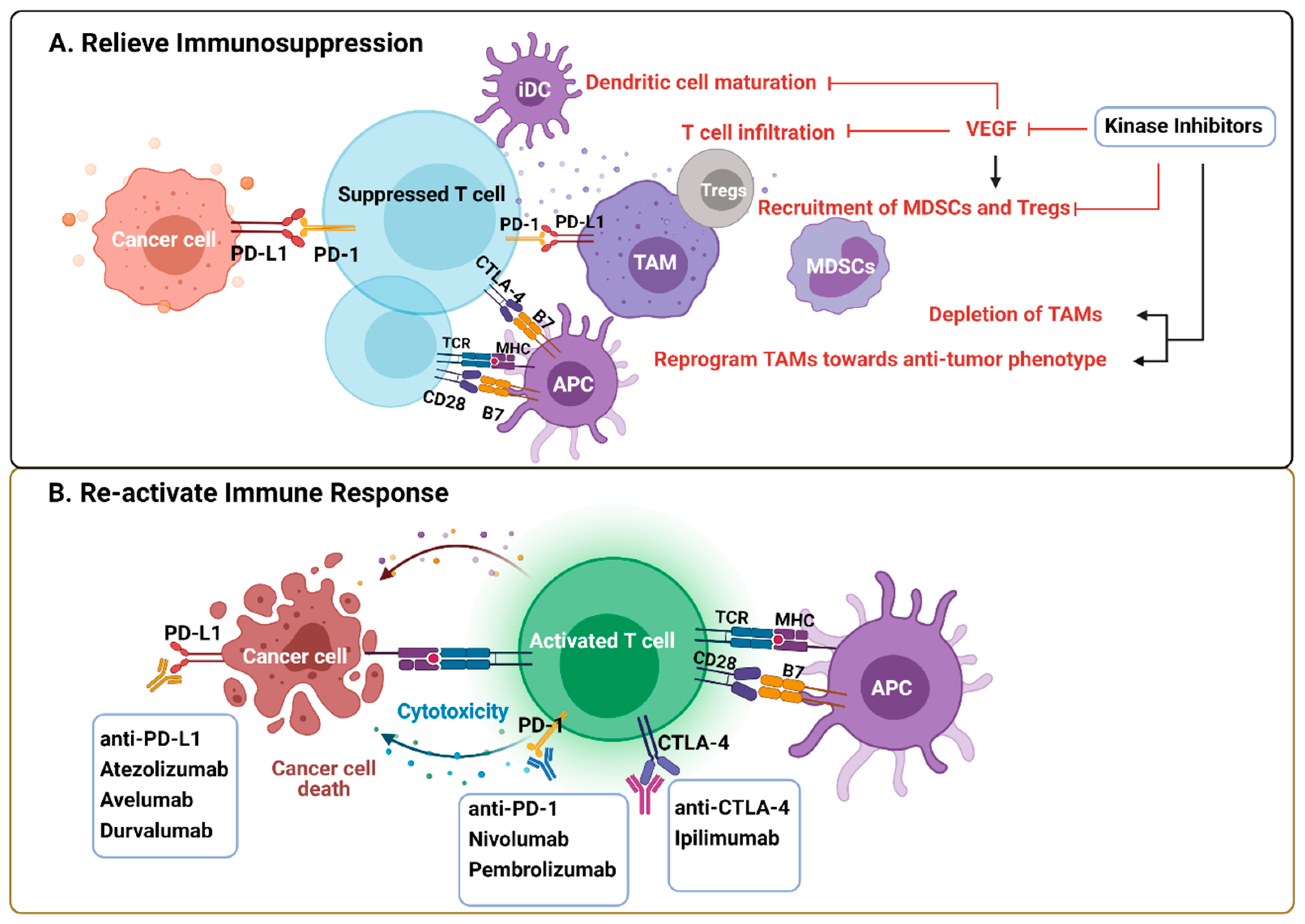
4. Immunological Actions of Kinase Inhibitors through Effects on Tumor Cells
5. Immunological Effects of VEGFR–MKIs
5.1. Immunologic Effects of VEGFR–MKIs Related to Angiogenic Alterations of the TME
5.2. Immunological Effects of VEGFR–MKIs Independent of the Angiogenic Pathway
6. Immunomodulation by Other Kinase Inhibitors
6.1. AXL Pathway
6.2. HGF/c–MET Axis
6.3. The Downstream MAPK Pathway and mTOR Pathway
7. Rationale of the Combined Therapy
8. Future Prospects/Remaining Questions
Author Contributions
Funding
Conflicts of Interest
References
- Thielen, N.; van der Holt, B.; Cornelissen, J.J.; Verhoef, G.E.; Gussinklo, T.; Biemond, B.J.; Daenen, S.M.; Deenik, W.; van Marwijk Kooy, R.; Petersen, E.; et al. Imatinib discontinuation in chronic phase myeloid leukaemia patients in sustained complete molecular response: A randomised trial of the Dutch-Belgian Cooperative Trial for Haemato-Oncology (HOVON). Eur. J. Cancer 2013, 49, 3242–3246. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Kato, Y.; Ozawa, Y.; Kodama, K.; Ito, J.; Ichikawa, K.; Yamada, K.; Hori, Y.; Tabata, K.; Takase, K.; et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018, 109, 3993–4002. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 11. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers 2020, 12, 3401. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Fakih, M.; Ouyang, C.; Wang, C.; Tu, T.Y.; Gozo, M.C.; Cho, M.; Sy, M.; Longmate, J.A.; Lee, P.P. Immune overdrive signature in colorectal tumor subset predicts poor clinical outcome. J. Clin. Investig. 2019, 129, 4464–4476. [Google Scholar] [CrossRef]
- Yagi, T.; Baba, Y.; Okadome, K.; Kiyozumi, Y.; Hiyoshi, Y.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Watanabe, M.; et al. Tumour-associated macrophages are associated with poor prognosis and programmed death ligand 1 expression in oesophageal cancer. Eur. J. Cancer 2019, 111, 38–49. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e1318. [Google Scholar] [CrossRef] [Green Version]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr.-Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Qing, W.; Fang, W.Y.; Ye, L.; Shen, L.Y.; Zhang, X.F.; Fei, X.C.; Chen, X.; Wang, W.Q.; Li, X.Y.; Xiao, J.C.; et al. Density of tumor-associated macrophages correlates with lymph node metastasis in papillary thyroid carcinoma. Thyroid 2012, 22, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e510. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassier, P.A.; Garin, G.; Eberst, L.; Delord, J.-P.; Chabaud, S.; Terret, C.; Montane, L.; Bidaux, A.-S.; Laurent, S.; Jaubert, L.; et al. MEDIPLEX: A phase 1 study of durvalumab (D) combined with pexidartinib (P) in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC). J. Clin. Oncol. 2019, 37, 2579. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavijo, P.E.; Friedman, J.; Robbins, Y.; Moore, E.C.; Smith, E.; Zauderer, M.; Evans, E.E.; Allen, C.T. Semaphorin4D Inhibition Improves Response to Immune-Checkpoint Blockade via Attenuation of MDSC Recruitment and Function. Cancer Immunol. Res. 2019, 7, 282–291. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Montor, W.R.; Salas, A.R.O.S.E.; de Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 20. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Zarakas, M.A.; Desai, J.V.; Chamilos, G.; Lionakis, M.S. Fungal Infections with Ibrutinib and Other Small-Molecule Kinase Inhibitors. Curr. Fungal Infect. Rep. 2019, 13, 86–98. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune Infiltrates Are Prognostic Factors in Localized Gastrointestinal Stromal Tumors. Cancer Res. 2013, 73, 3499. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Pol, J.; Levesque, S.; Petrazzuolo, A.; Pfirschke, C.; Engblom, C.; Rickelt, S.; Yamazaki, T.; Iribarren, K.; et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 2019, 10, 1486. [Google Scholar] [CrossRef]
- Gunda, V.; Gigliotti, B.; Ashry, T.; Ndishabandi, D.; McCarthy, M.; Zhou, Z.; Amin, S.; Lee, K.E.; Stork, T.; Wirth, L.; et al. Anti-PD-1/PD-L1 therapy augments lenvatinib’s efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int. J. Cancer 2019, 144, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Luo, H.; Shi, B.Z.; Di, S.M.; Sun, R.X.; Su, J.W.; Liu, Y.; Li, H.; Jiang, H.; Li, Z.H. Combined Antitumor Effects of Sorafenib and GPC3-CAR T Cells in Mouse Models of Hepatocellular Carcinoma. Mol. Ther. 2019, 27, 1483–1494. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.Y.; Peng, Y.J.; Zhao, S.Y.; Mou, J.; Zeng, L.S.; Jiang, X.H.; Yang, C.L.; Huang, C.; Li, Y.Y.; Lu, Y.; et al. Combination Foretinib and Anti-PD-1 Antibody Immunotherapy for Colorectal Carcinoma. Front. Cell Dev. Biol. 2021, 9, 16. [Google Scholar] [CrossRef]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Ou, D.-L.; Chen, C.-W.; Hsu, C.-L.; Chung, C.-H.; Feng, Z.-R.; Lee, B.-S.; Cheng, A.-L.; Yang, M.-H.; Hsu, C. Regorafenib enhances antitumor immunity via inhibition of p38 kinase/Creb1/Klf4 axis in tumor-associated macrophages. J. Immunother. Cancer 2021, 9, e001657. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728. [Google Scholar] [CrossRef] [Green Version]
- Poon, E.; Mullins, S.; Watkins, A.; Williams, G.S.; Koopmann, J.O.; Di Genova, G.; Cumberbatch, M.; Veldman-Jones, M.; Grosskurth, S.E.; Sah, V.; et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J. Immunother. Cancer 2017, 5, 14. [Google Scholar] [CrossRef]
- Draghiciu, O.; Nijman, H.W.; Hoogeboom, B.N.; Meijerhof, T.; Daemen, T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology 2015, 4, e989764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.C.; Lee, Y.H.; Chang, C.J.; Shun, C.T.; Fang, C.Y.; Shao, Y.Y.; Liu, T.H.; Cheng, A.L.; Hsu, C.H. Increased Expression of Programmed Death-Ligand 1 in Infiltrating Immune Cells in Hepatocellular Carcinoma Tissues after Sorafenib Treatment. Liver Cancer 2019, 8, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.I.; Chaput, N.; Poirier-Colame, V.; Rusakiewicz, S.; Jacquelot, N.; Chaba, K.; Mortier, E.; Jacques, Y.; Caillat-Zucman, S.; Flament, C.; et al. Regulation of CD4(+)NKG2D(+) Th1 cells in patients with metastatic melanoma treated with sorafenib: Role of IL-15Rα and NKG2D triggering. Cancer Res. 2014, 74, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Sprinzl, M.F.; Reisinger, F.; Puschnik, A.; Ringelhan, M.; Ackermann, K.; Hartmann, D.; Schiemann, M.; Weinmann, A.; Galle, P.R.; Schuchmann, M.; et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology 2013, 57, 2358–2368. [Google Scholar] [CrossRef]
- Heine, A.; Schilling, J.; Grünwald, B.; Krüger, A.; Gevensleben, H.; Held, S.A.; Garbi, N.; Kurts, C.; Brossart, P.; Knolle, P.; et al. The induction of human myeloid derived suppressor cells through hepatic stellate cells is dose-dependently inhibited by the tyrosine kinase inhibitors nilotinib, dasatinib and sorafenib, but not sunitinib. Cancer Immunol. Immunother. 2016, 65, 273–282. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, Y.H.; Chiu, C.J.; Lu, L.C.; Liao, C.C.; Liang, C.W.; Hsu, C.H.; Cheng, A.L. Targeting tumor-infiltrating Ly6G(+) myeloid cells improves sorafenib efficacy in mouse orthotopic hepatocellular carcinoma. Int. J. Cancer 2018, 142, 1878–1889. [Google Scholar] [CrossRef] [Green Version]
- Hage, C.; Hoves, S.; Strauss, L.; Bissinger, S.; Prinz, Y.; Pöschinger, T.; Kiessling, F.; Ries, C.H. Sorafenib Induces Pyroptosis in Macrophages and Triggers Natural Killer Cell-Mediated Cytotoxicity Against Hepatocellular Carcinoma. Hepatology 2019, 70, 1280–1297. [Google Scholar] [CrossRef]
- Alfaro, C.; Suarez, N.; Gonzalez, A.; Solano, S.; Erro, L.; Dubrot, J.; Palazon, A.; Hervas-Stubbs, S.; Gurpide, A.; Lopez-Picazo, J.M.; et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br. J. Cancer 2009, 100, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, I.G.; Napoletano, C.; Botticelli, A.; Caponnetto, S.; Calabrò, F.; Gelibter, A.; Rughetti, A.; Ruscito, I.; Rahimi, H.; Rossi, E.; et al. TK Inhibitor Pazopanib Primes DCs by Downregulation of the β-Catenin Pathway. Cancer Immunol. Res. 2018, 6, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Swanson, K.D.; Csizmadia, E.; Solanki, A.; Landon-Brace, N.; Gehring, M.P.; Helenius, K.; Olson, B.M.; Pyzer, A.R.; Wang, L.C.; et al. Cabozantinib Eradicates Advanced Murine Prostate Cancer by Activating Antitumor Innate Immunity. Cancer Discov. 2017, 7, 750–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658. [Google Scholar] [CrossRef] [Green Version]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.R.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349. [Google Scholar] [CrossRef]
- Garg, A.D.; Romano, E.; Rufo, N.; Agostinis, P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: Mechanisms and clinical translation. Cell Death Differ. 2016, 23, 938–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Sawano, A.; Iwai, S.; Sakurai, Y.; Ito, M.; Shitara, K.; Nakahata, T.; Shibuya, M. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood 2001, 97, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Osada, T.; Chong, G.; Tansik, R.; Hong, T.; Spector, N.; Kumar, R.; Hurwitz, H.I.; Dev, I.; Nixon, A.B.; Lyerly, H.K.; et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 2008, 57, 1115–1124. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Farsaci, B.; Donahue, R.N.; Coplin, M.A.; Grenga, I.; Lepone, L.M.; Molinolo, A.A.; Hodge, J.W. Immune consequences of decreasing tumor vasculature with antiangiogenic tyrosine kinase inhibitors in combination with therapeutic vaccines. Cancer Immunol. Res. 2014, 2, 1090–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassara, A.; Rmili, C.W.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef]
- Chen, M.L.; Yan, B.S.; Lu, W.C.; Chen, M.H.; Yu, S.L.; Yang, P.C.; Cheng, A.L. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int. J. Cancer 2014, 134, 319–331. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petroni, G.; Buque, A.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Immunomodulation by targeted anticancer agents. Cancer Cell 2021, 39, 310–345. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Aehnlich, P.; Powell, R.M.; Peeters, M.J.W.; Rahbech, A.; Thor Straten, P. TAM Receptor Inhibition-Implications for Cancer and the Immune System. Cancers 2021, 13, 1195. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.Y.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Bergerot, P.; Lamb, P.; Wang, E.; Pal, S.K. Cabozantinib in Combination with Immunotherapy for Advanced Renal Cell Carcinoma and Urothelial Carcinoma: Rationale and Clinical Evidence. Mol. Cancer 2019, 18, 2185–2193. [Google Scholar] [CrossRef] [Green Version]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, F.; Giovannetti, E.; Peters, G.J.; Firuzi, O. Combination of HGF/MET-targeting agents and other therapeutic strategies in cancer. Crit. Rev. Oncol. Hematol. 2021, 160, 103234. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Allegrezza, M.J.; Rutkowski, M.R.; Stephen, T.L.; Svoronos, N.; Perales-Puchalt, A.; Nguyen, J.M.; Payne, K.K.; Singhal, S.; Eruslanov, E.B.; Tchou, J.; et al. Trametinib Drives T-cell-Dependent Control of KRAS-Mutated Tumors by Inhibiting Pathological Myelopoiesis. Cancer Res. 2016, 76, 6253–6265. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.; Bex, A.; et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1241–1250. [Google Scholar] [CrossRef]
- Granito, A.; Marinelli, S.; Forgione, A.; Renzulli, M.; Benevento, F.; Piscaglia, F.; Tovoli, F. Regorafenib Combined with Other Systemic Therapies: Exploring Promising Therapeutic Combinations in HCC. J. Hepatocell. Carcinoma 2021, 8, 477–492. [Google Scholar] [CrossRef]
- Rini, B.I.; Atkins, M.B.; Plimack, E.R.; Soulières, D.; McDermott, R.S.; Bedke, J.; Tartas, S.; Alekseev, B.; Melichar, B.; Shparyk, Y.; et al. Characterization and Management of Treatment-emergent Hepatic Toxicity in Patients with Advanced Renal Cell Carcinoma Receiving First-line Pembrolizumab plus Axitinib. Results from the KEYNOTE-426 Trial. Eur. Urol. Oncol. 2021. [Google Scholar] [CrossRef]
- Chowdhury, S.; McDermott, D.F.; Voss, M.H.; Hawkins, R.E.; Aimone, P.; Voi, M.; Isabelle, N.; Wu, Y.; Infante, J.R. A phase I/II study to assess the safety and efficacy of pazopanib (PAZ) and pembrolizumab (PEM) in patients (pts) with advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2017, 35, 4506. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Bedke, J.; Albiges, L.; Capitanio, U.; Giles, R.H.; Hora, M.; Lam, T.B.; Ljungberg, B.; Marconi, L.; Klatte, T.; Volpe, A.; et al. The 2021 Updated European Association of Urology Guidelines on Renal Cell Carcinoma: Immune Checkpoint Inhibitor-based Combination Therapies for Treatment-naive Metastatic Clear-cell Renal Cell Carcinoma Are Standard of Care. Eur. Urol. 2021, 80, 393–397. [Google Scholar] [CrossRef]
- Huelse, J.M.; Fridlyand, D.M.; Earp, S.; DeRyckere, D.; Graham, D.K. MERTK in cancer therapy: Targeting the receptor tyrosine kinase in tumor cells and the immune system. Pharmacol. Ther. 2020, 213, 107577. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Denny, W.A.; Flanagan, J.U. Small-molecule CSF1R kinase inhibitors; review of patents 2015-present. Expert Opin. Ther. Pat. 2021, 31, 107–117. [Google Scholar] [CrossRef]
- Priem, B.; van Leent, M.M.T.; Teunissen, A.J.P.; Sofias, A.M.; Mourits, V.P.; Willemsen, L.; Klein, E.D.; Oosterwijk, R.S.; Meerwaldt, A.E.; Munitz, J.; et al. Trained Immunity-Promoting Nanobiologic Therapy Suppresses Tumor Growth and Potentiates Checkpoint Inhibition. Cell 2020, 183, 786–801.e719. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Targets | Clinical Indications | Kinase Inhibitors |
|---|---|---|
| Receptor Tyrosine Kinase | ||
| VEGFR (multi-targeted) | RCC, HCC, GIST, DTC, MTC, CRC | Sorafenib (2005), Sunitinib (2006), Pazopanib (2009), Vandetanib (2011), Axitinib (2011), Cabozantinib (2012), Regorafenib (2012), Lenvatinb (2015), Tivozanib (2021) |
| EGFR | Lung cancer | Gefitinib (2002), Erlotinib (2004), Afatinib (2013), Osimetinib (2015), Olmutinib (2016), Brigatinib (2017), Dacomitinib (2018) |
| ALK | Lung cancer | Crizotinib (2011), Ceritinib (2014), Alectinib (2014), Lorlatinib (2018) |
| MET | Lung cancer | Tepotinib (2019), Capmatinib (2020) |
| RET | Lung, thyroid cancer | Selpercatinib (2020), Pralsetinib (2020) |
| Her2 | Breast Cancer | Lapatinib (2007), Neratinib (2017), Tucatinib (2020) |
| Kit, PDGFR, CSF1R, FLT3 | GIST | Ripretinib (2020), Avapritinib (2020) |
| TGCT | Pexidartinib (2019) | |
| FGFR | Bladder, cholangiocarcinoma | Erdafitinib (2019), Pemigatinib (2020) |
| Trk | Any metastatic solid tumor with NTRK mutations | Larotrectinib (2018), Entrectinib (2019) |
| Serine/threonine Protein Kinase | ||
| CDK family | Breast cancer | Palbociclib (2015), Ribociclib (2017) |
| B-Raf, MEK1/2 pathway | Melanoma | Vemurafenib (2011), Dabrafenib (2013), Trametinib (2013), Cobimetinib (2015), Encorafenib (2018), Binimetinib (2018) |
| Neurofibromatosis type I | Selumetinib (2020) | |
| PI3K pathway | Breast Cancer | Alpelisib (2019) |
| Impact on Immune Cells | Drug | Tumor Type | Model | Impact on Tumor Cells | Combination with Immunotherapies | Efficacy of Combination with Immunotherapy | Ref. |
|---|---|---|---|---|---|---|---|
| Increases proinflammatory Mφ and T cell infiltration, Upregulates PD-1 and CTLA-4 expression | Crizotinib 10µM (plus cisplatin) | NSCLC | Transplantable tumor in immunocompetent murine model | Induces ICD, Upregulates PD-L1 expression | Anti-PD-1 Anti-CTLA-4 | Reaches 100% cure rate | [34] |
| Decreases tumor-infiltrating monocytes and macrophages. Increases CD8+ T cells | Lenvatinib | HCC | Immunocompetent murine model (compared to immunocompromised) | Anti-PD-1 | Improves tumor regression | [2] | |
| Increases tumor-infiltrating macrophages, CD8+ T-cells, Tregs, PMN-MDSCs | Lenvatinib | ATC | Orthotopic tumor murine model | Anti-PD-1 | Improves tumor reduction | [35] | |
| Increases IL12 secretion from TAMs | Sorafenib | HCC | Immunocompetent murine model (compared to immunocompromised) | Induces cancer cell apoptosis | GPC3-targeted chimeric antigen receptor T cell therapy | Increases animal survival | [36] |
| Decreases TAMs, Promotes T cells infiltration | Foretinib | Colorectal Carcinoma | Subcutaneous tumor in murine model | Increases PD-L1 expression on tumor cells | Anti-PD-1 | Improves tumor regression, prolongs overall survival | [37] |
| Reduces TAMs and MDSCs in TME Promotes T cell expansion | BRAF inhibitors + MEK inhibitors | Melanoma | Immunocompetent murine model (compared with immunocompromised model) | Induces pyroptosis | ND | [38] | |
| Reprograms Mφ towards anti-tumor phenotype | Regorafenib | HCC | In vitro | Anti-PD1 | Reduces tumor growth, Improves animal survival | [39] | |
| Selectively depletes MDSCs | Cabozantinib, BEZ235 | Prostate cancer | Spontaneous tumor in immunocompetent murine model | Inhibits PI3K pathway; Reduces CCL5, CCL12, CD40, HGF, Increases IL-1ra, CD142, and VEGF released by tumor cells | Anti-CTLA-4 + Anti-PD-1 | Decreases tumor size, Reduces lymph node metastasis and lung micro-metastasis | [40] |
| Decreases infiltration of granulocytic MDSCs and neutrophils | Selumetinib | Colorectal | Transplantable tumor in murine model | Anti-CTLA-4 | Reduces tumor volume, Prolongs animal survival | [41] | |
| Depletes MDSCs Increases CD8+ T cells | Sunitinib | HPV-Induced cancer | Induced tumor in murine model | Cancer vaccine | Increases survival rate | [42] | |
| Impairs recruitment of immunosuppressive TAN, enhances T cell expansion | Capmatinib | Melanoma Lung Breast Colon | Transplantable tumor + Inducible primary tumor in murine model | Anti-PD-1, PCP | Increases animal survival | [43] | |
| Increases PD-L1 expression in TAMs | Sorafenib | HCC | Patient tissue | ND | [44] | ||
| Upregulates IL-15Ra expression on circulating monocytes | Sorafenib | Melanoma | Patient tissue | ND | [45] | ||
| Favors the expansion of proinflammatory TAMs, activates antitumor response of NKs | Sorafenib | HCC | Murine HCC model + In vitro | ND | [46] | ||
| Negatively affects the differentiation of monocytes into functional MDSCs | Sorafenib | NA | In vitro | ND | [47] | ||
| Increases MDSCs infiltration | Sorafenib | HCC | Orthotopic liver or subcutaneous tumor in murine model | ND | [48] | ||
| Induces Mφ pyroptosis, activates NK cells anti-tumor response | Sorafenib | HCC | Spontaneous and transplantable murine model + in vitro | ND | [49] | ||
| Restores DCs maturation | Sorafenib | NA | In vitro | ND | [50] | ||
| Improves DCs differentiation and performance | Pazopanib | NA | In vitro | ND | [51] | ||
| Increases neutrophil infiltration and anti-tumor activity | Cabozantinib | Prostate cancer | Genetic engineered tumor murine model | Triggers release of CXCL12 and HMGB1 from dying tumor cells | ND | [52] | |
| Increases TANs infiltration | Sorafenib | HCC | Transplantable murine model + patient tissue biopsy | ND | [53] | ||
| Reduces anti-tumor TANs recruitment | MET TKIs (PF-04217903, INCB28060, JNJ-38877605) | Melanoma | Transplantable tumor in Met conditional knockout mice | ND | [54] |
| Immunotherapy | Kinase Inhibitor Treatment | Conditions | Enrollment (Estimated) | NCT Number | Outcome Measures |
|---|---|---|---|---|---|
| Pembrolizumab (PD-1) | Lenvatinib | Endometrial Neoplasms | 827 | NCT03517449 | OS, PFS |
| HCC | 750 | NCT03713593 | OS, PFS | ||
| Malignant Melanoma | 660 | NCT03820986 | OS, PFS | ||
| Nonsquamous NSCLC | 726 | NCT03829319 | OS, PFS | ||
| NSCLC | 620 | NCT03829332 | OS, PFS | ||
| Endometrial Neoplasms | 875 | NCT03884101 | OS, PFS | ||
| Urothelial Carcinoma | 694 | NCT03898180 | OS, PFS | ||
| Metastatic NSCLC | 405 | NCT03976375 | OS, PFS | ||
| HNSCC | 500 | NCT04199104 | OS, PFS, ORR | ||
| HCC | 950 | NCT04246177 | OS, PFS | ||
| Advanced/Metastatic GEA | 790 | NCT04662710 | OS, PFS | ||
| RCC | 1431 | NCT04736706 | OS, PFS | ||
| CRC | 434 | NCT04776148 | OS | ||
| Advanced/Metastatic RCC | 1069 | NCT02811861 * | Prolonged PFS (23.9 vs. 9.2 m) Improves OS ** (HR, 0.66) | ||
| Pembrolizumab | Axitinib | Advanced/Metastatic RCC | 861 | NCT02853331 * | Prolonged PFS (15.1 vs. 11.1 m) Improves OS (89.9% vs. 78.3%) Increases ORR (59.3% vs. 35.7%) |
| Pembrolizumab | Encorafenib | Melanoma | 624 | NCT04657991 | PFS |
| Atezolizumab (PD-L1) | Cabozantinib | HCC | 740 | NCT03755791 | OS, PFS |
| RCC | 500 | NCT04338269 | OS, PFS | ||
| Metastatic Prostate Cancer | 580 | NCT04446117 | OS, PFS | ||
| NSCLC | 350 | NCT04471428 | OS | ||
| Atezolizumab | Sorafenib/Lenvatinib | Unresectable HCC | 554 | NCT04770896 | OS |
| Nivolumab (PD-1)/Ipilimumab (CTLA-4) | Cabozantinib | Metastatic RCC | 1046 | NCT03793166 | OS |
| RCC | 840 | NCT03937219 | PFS | ||
| Nivolumab (PD-1) | Cabozantinib | Advanced/Metastatic RCC | 638 | NCT03141177 * | Prolonged PFS (16.6 vs. 8.3 m) Increases OS (85.7% vs. 75.6%) Increases ORR (55.7% vs. 27.1%) |
| Nivolumab (PD-1) | Sitravatinib | Metastatic Non-Squamous NSCLC | 532 | NCT03906071 | OS |
| Avelumab (PD-L1) | Axitinib | RCC | 888 | NCT02684006 | OS, PFS |
| IMA901 (cancer vaccine) | Sunitinib | Metastatic RCC | 339 | NCT01265901 | OS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, C.; Rabold, K.; Mulder, W.J.M.; Jaeger, M.; Netea-Maier, R.T. Kinase Inhibitors’ Effects on Innate Immunity in Solid Cancers. Cancers 2021, 13, 5695. https://doi.org/10.3390/cancers13225695
Peng C, Rabold K, Mulder WJM, Jaeger M, Netea-Maier RT. Kinase Inhibitors’ Effects on Innate Immunity in Solid Cancers. Cancers. 2021; 13(22):5695. https://doi.org/10.3390/cancers13225695
Chicago/Turabian StylePeng, Chunying, Katrin Rabold, Willem J. M. Mulder, Martin Jaeger, and Romana T. Netea-Maier. 2021. "Kinase Inhibitors’ Effects on Innate Immunity in Solid Cancers" Cancers 13, no. 22: 5695. https://doi.org/10.3390/cancers13225695
APA StylePeng, C., Rabold, K., Mulder, W. J. M., Jaeger, M., & Netea-Maier, R. T. (2021). Kinase Inhibitors’ Effects on Innate Immunity in Solid Cancers. Cancers, 13(22), 5695. https://doi.org/10.3390/cancers13225695