
Anti-Tumor Activity of Expanded PBMC-Derived NK Cells by Feeder-Free Protocol in Ovarian Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Mice
2.2. NK Cell Expansion Ex Vivo
2.3. Analysis of Receptors Expression Level of eNKs
2.4. Antibody
2.5. eNKs Cytotoxicity Assay
2.5.1. Flow-Based Killing Assay
2.5.2. RTCA-Based Killing Assay
2.6. Analysis of the Distribution of eNKs in NCG Mice
2.7. Tumor Mouse Models
2.8. Histopathological Evaluation and Blood Analysis
2.9. Statistical Analysis
3. Results
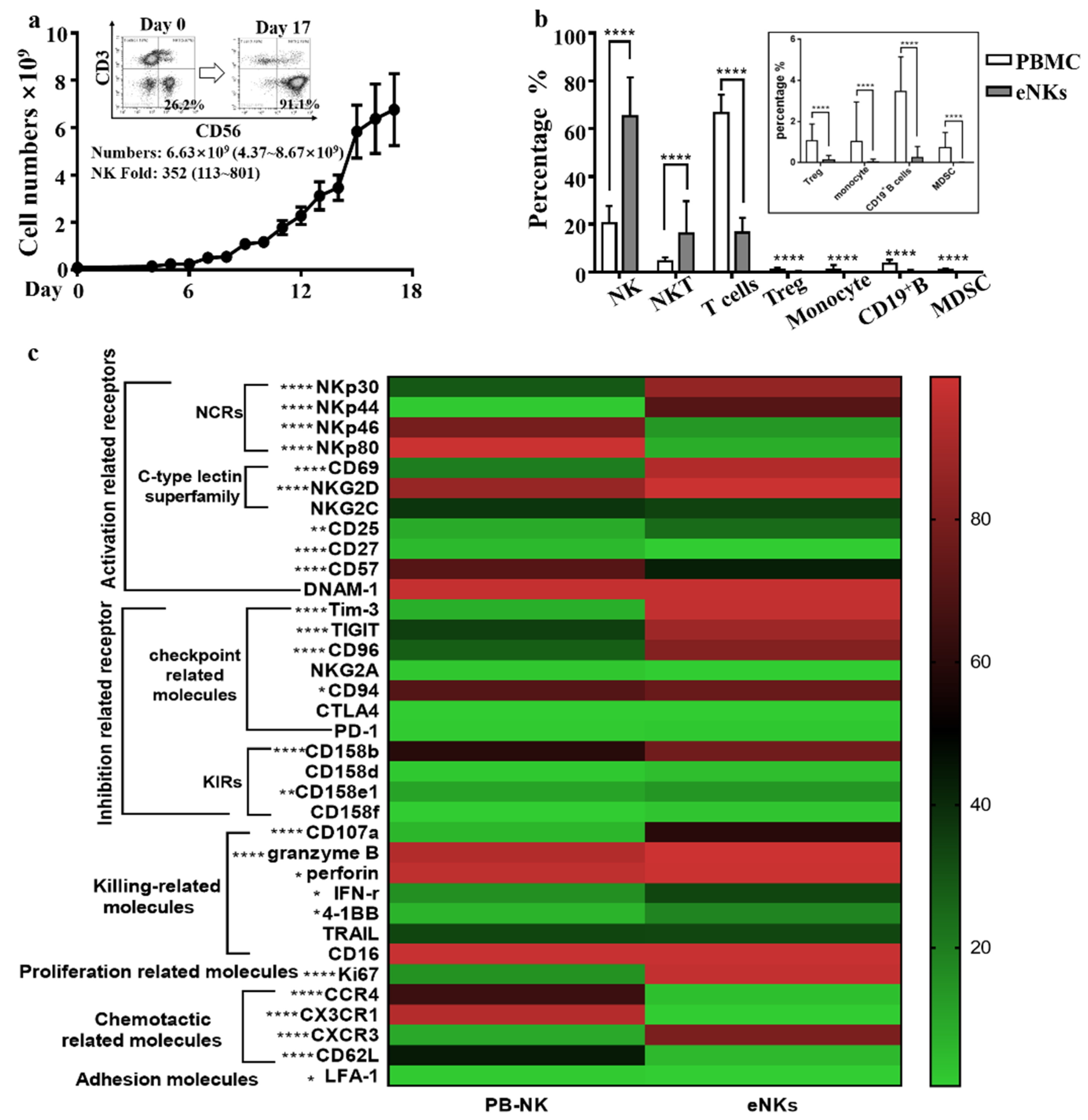
3.1. Expansion of PBMC-NK Ex Vivo Yields a High Quantity of High Purity NK Cells with Enhanced Cytotoxicity
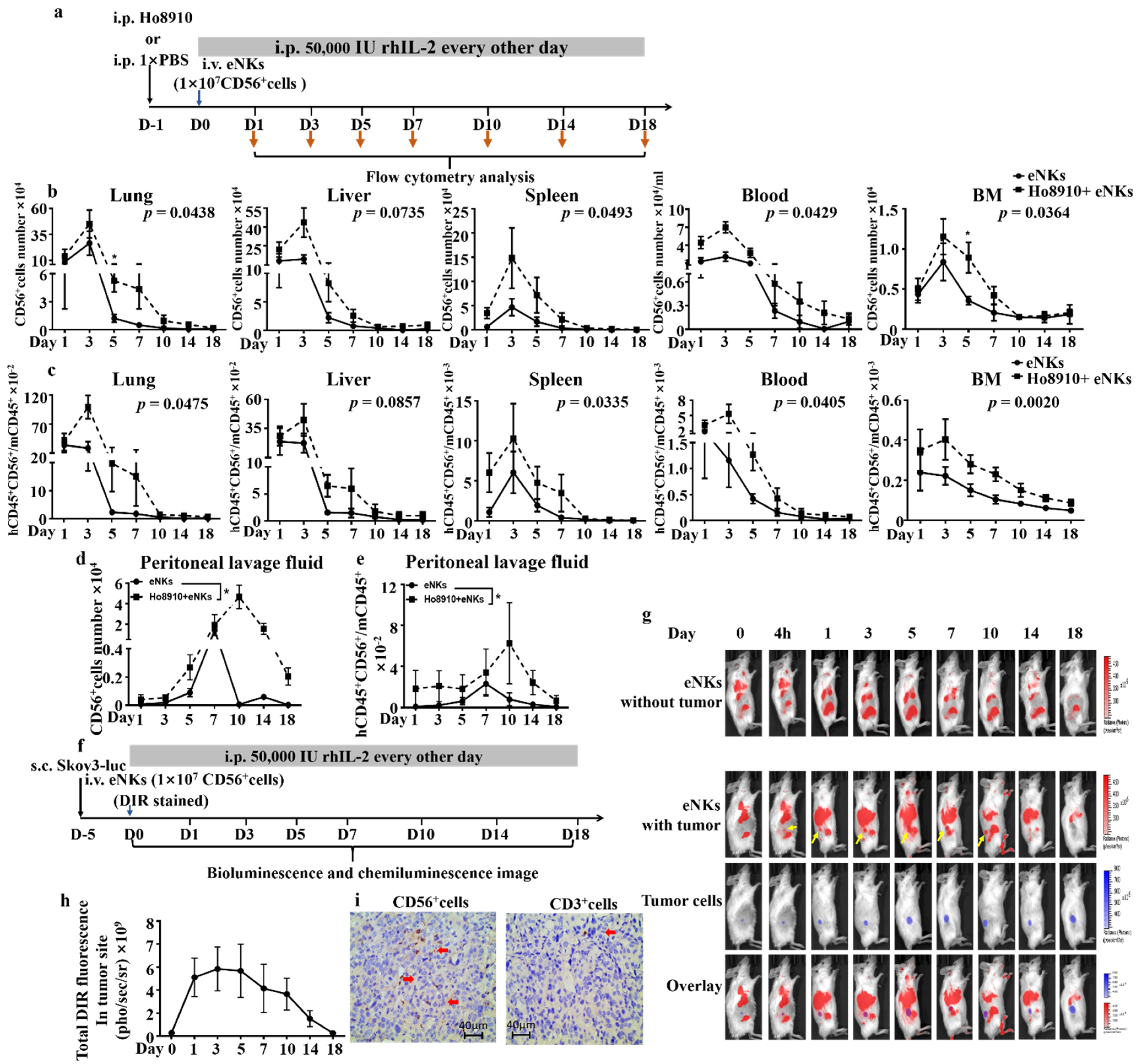
3.2. Proliferation, Lifespan, and Tumor Co-Localization Were Enhanced in eNKs in Ovarian Tumor-Bearing Mice
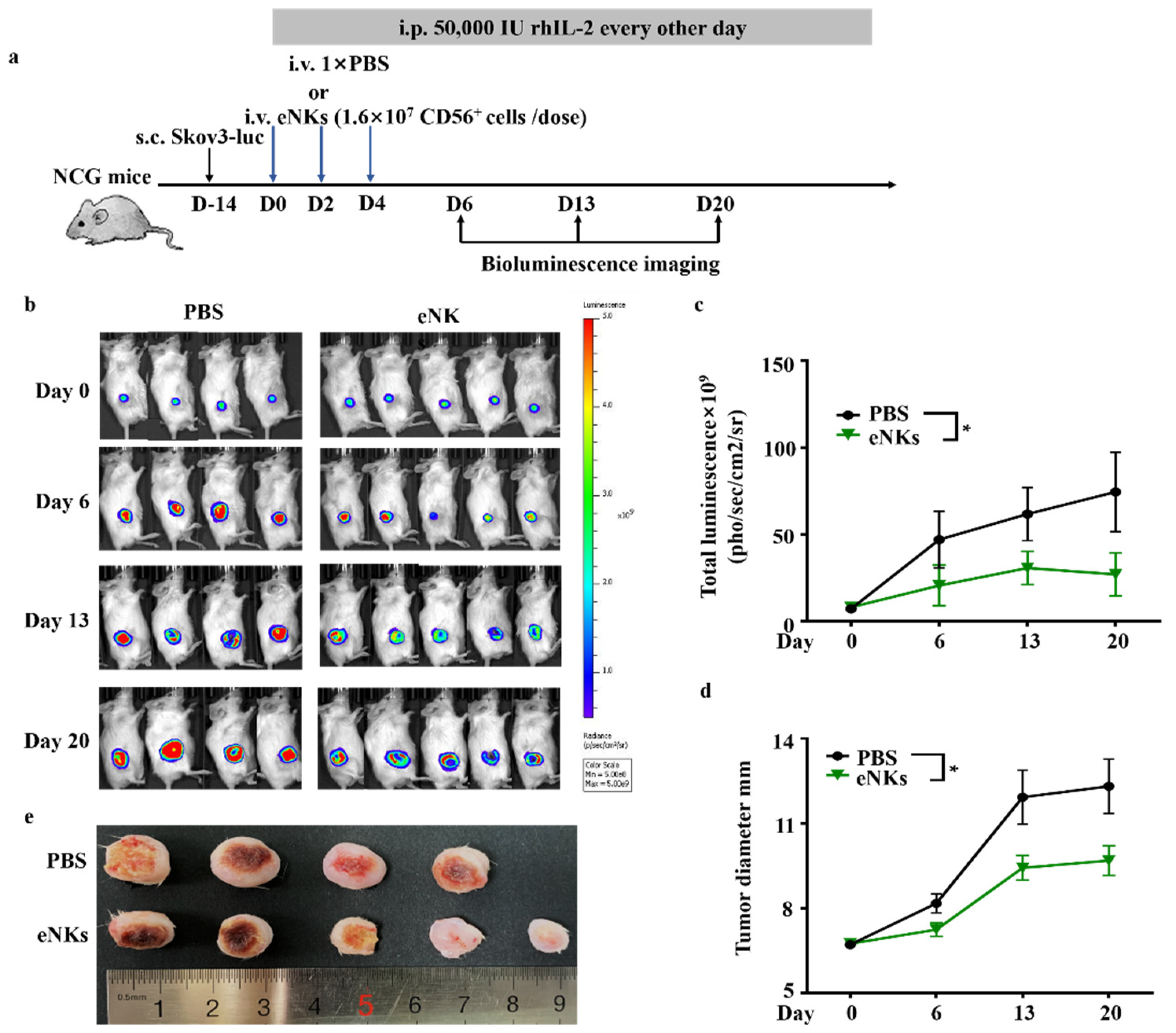
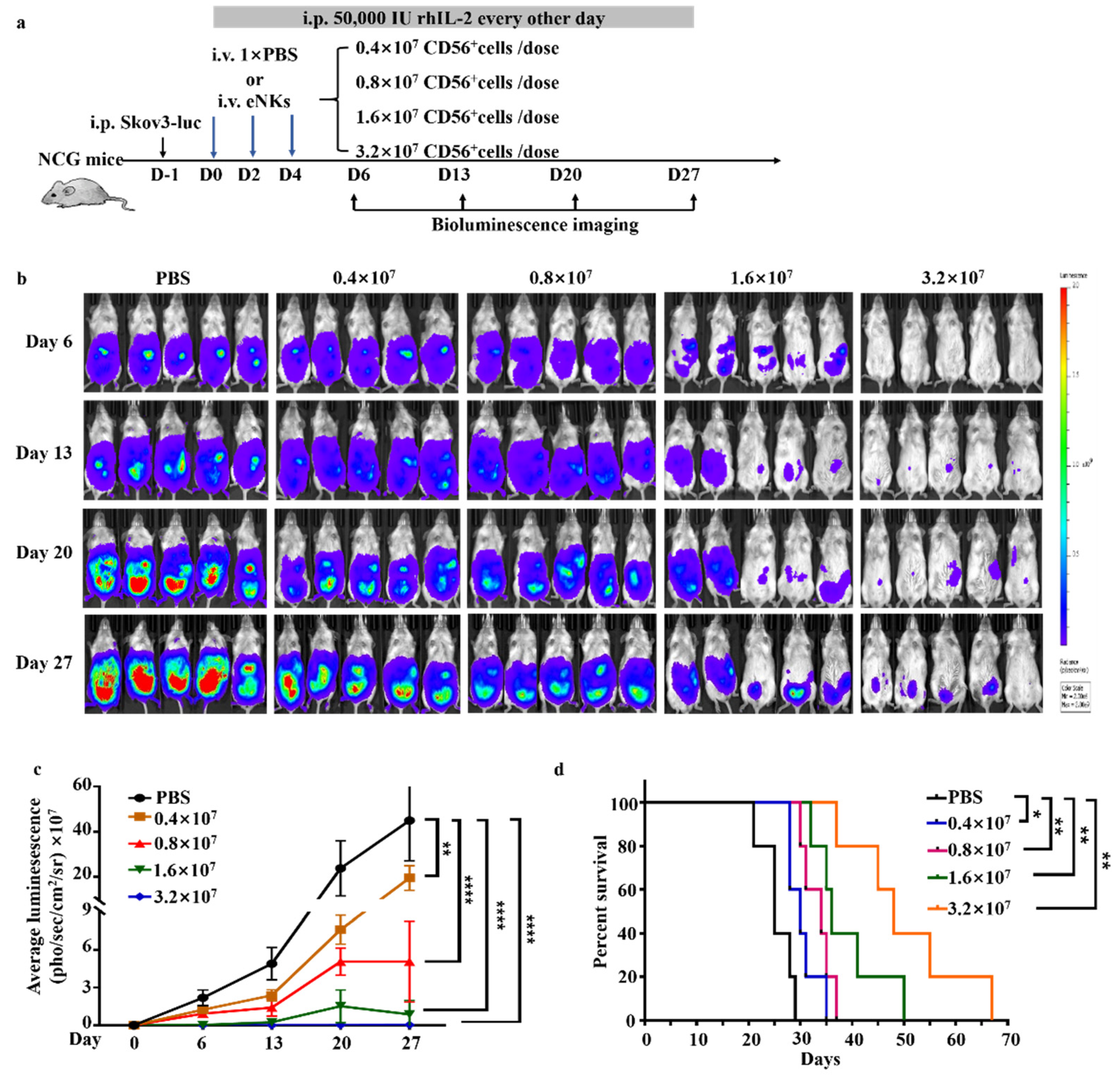
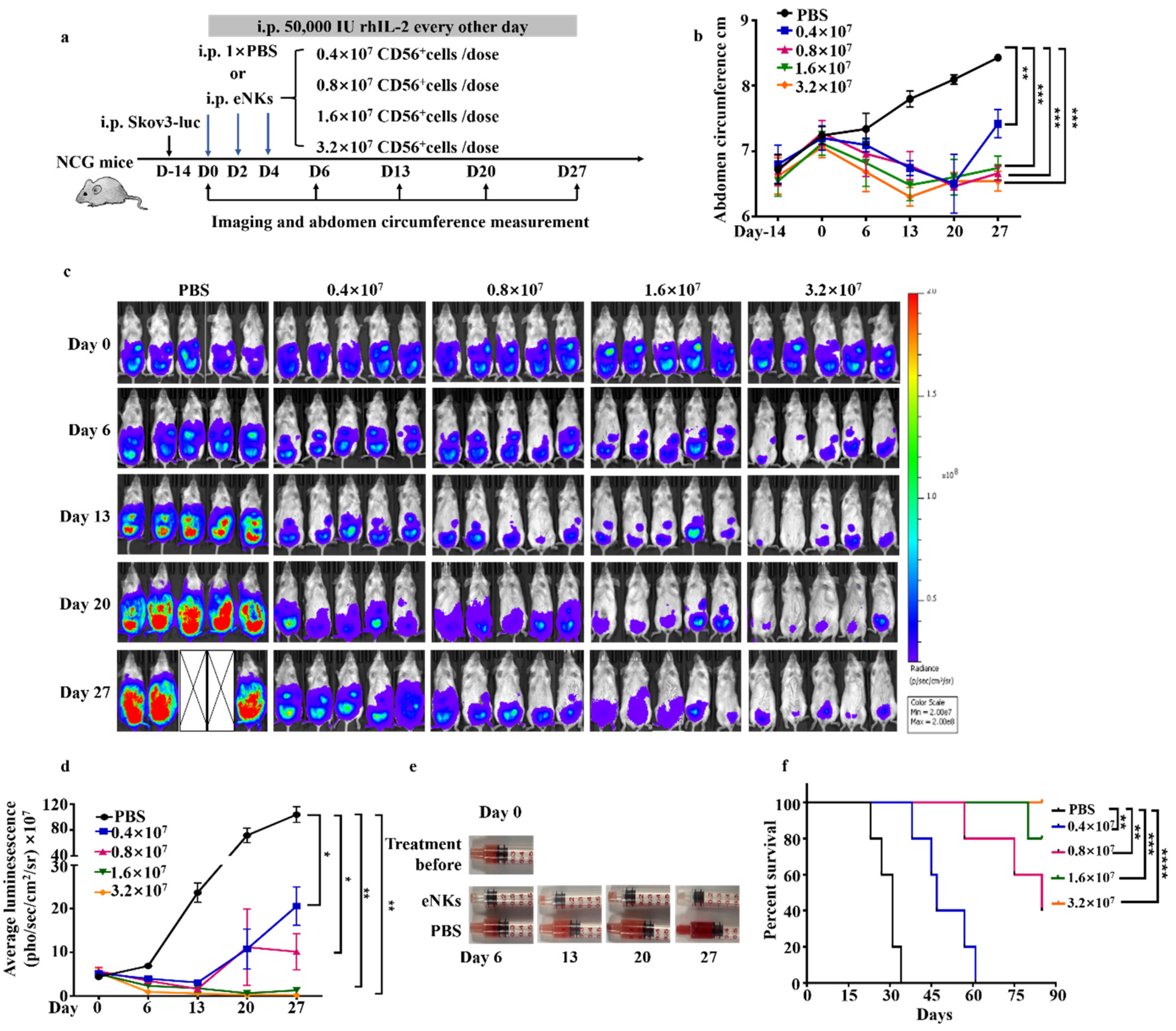
3.3. eNKs Effectively Reduced Ovarian Ascites and Tumor Burden in Ovarian Tumor-Bearing Mice
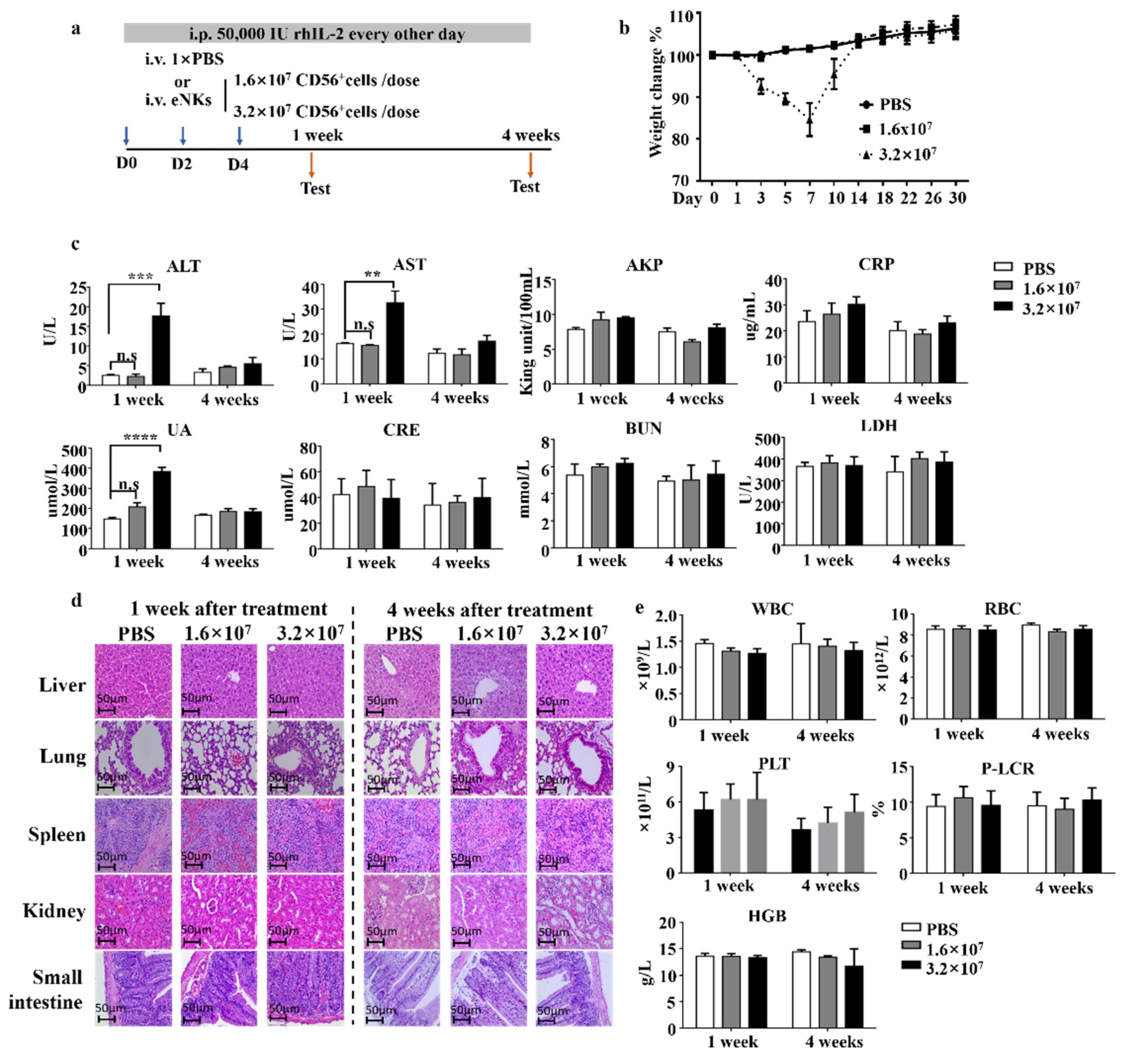
3.4. Safety Evaluation of eNKs In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science 2011, 331, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Navin, I.; Lam, M.T.; Parihar, R. Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment. Cancers 2020, 12, 3871. [Google Scholar] [CrossRef]
- Desbois, M.; Rusakiewicz, S.; Locher, C.; Zitvogel, L.; Chaput, N. Natural killer cells in non-hematopoietic malignancies. Front. Immunol. 2012, 3, 395. [Google Scholar] [CrossRef] [Green Version]
- Szmania, S.; Lapteva, N.; Garg, T.; Greenway, A.; Lingo, J.; Nair, B.; Stone, K.; Woods, E.; Khan, J.; Stivers, J.; et al. Ex Vivo-expanded Natural Killer Cells Demonstrate Robust Proliferation In Vivo in High-risk Relapsed Multiple Myeloma Patients. J. Immunother. 2015, 38, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Bachanova, V.; Sarhan, D.; DeFor, T.E.; Cooley, S.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Curtsinger, J.M.; Burns, L.; Weisdorf, D.J.; Miller, J.S. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol. Immunother. 2018, 67, 483–494. [Google Scholar] [CrossRef]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Lo, H.C.; Xu, Z.; Kim, I.S.; Pingel, B.; Aguirre, S.; Kodali, S.; Liu, J.; Zhang, W.; Muscarella, A.M.; Hein, S.M.; et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat. Cancer 2020, 1, 709–722. [Google Scholar] [CrossRef]
- Chen, K.-G.; Liu, A.; Jiang, C.-T.; Zhao, D.-K.; Ye, Q.-N.; Liao, Y.-Q.; Xu, C.-F.; Shen, S.; Wang, J. Dual-functional super bispecific nano-antibodies derived from monoclonal antibodies potentiate the antitumor effect of innate immune cells. Nano Today 2021, 39, 101209. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Nham, T.; Chew, M.V.; Lee, A.J.; Hammill, J.A.; Fan, I.Y.; Butcher, M.; Bramson, J.L.; Lee, D.A.; Hirte, H.W.; et al. Expanded CD56superbrightCD16+ NK Cells from Ovarian Cancer Patients Are Cytotoxic against Autologous Tumor in a Patient-Derived Xenograft Murine Model. Cancer Immunol. Res. 2018, 6, 1174–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Reader, J.; Roque, D.M. Review of Immune Therapies Targeting Ovarian Cancer. Curr. Treat. Opt. Oncol. 2018, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, C.V.; Tetlow, A.L.; Bantis, L.E.; Godwin, A.K. Reducing Ovarian Cancer Mortality through Early Detection: Approaches Using Circulating Biomarkers. Cancer Prev. Res. 2020, 13, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Hoogstad-van Evert, J.S.; Cany, J.; van den Brand, D.; Oudenampsen, M.; Brock, R.; Torensma, R.; Bekkers, R.L.; Jansen, J.H.; Massuger, L.F.; Dolstra, H. Umbilical cord blood CD34+ progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rgnull mice. Oncoimmunology 2017, 6, e1320630. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.; Matte, I.; Garde-Granger, P.; Bessette, P.; Piche, A. Ascites IL-10 Promotes Ovarian Cancer Cell Migration. Cancer Microenviron. 2018, 11, 115–124. [Google Scholar] [CrossRef]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [Green Version]
- Krugmann, J.; Schwarz, C.L.; Melcher, B.; Sterlacci, W.; Ozalinskaite, A.; Lermann, J.; Agaimy, A.; Vieth, M. Malignant ascites occurs most often in patients with high-grade serous papillary ovarian cancer at initial diagnosis: A retrospective analysis of 191 women treated at Bayreuth Hospital, 2006–2015. Arch. Gynecol. Obstet. 2019, 299, 515–523. [Google Scholar] [CrossRef]
- Nham, T.; Poznanski, S.M.; Fan, I.Y.; Shenouda, M.M.; Chew, M.V.; Lee, A.J.; Vahedi, F.; Karimi, Y.; Butcher, M.; Lee, D.A.; et al. Ex vivo-expanded NK cells from blood and ascites of ovarian cancer patients are cytotoxic against autologous primary ovarian cancer cells. Cancer Immunol. Immunother. 2018, 67, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Kwon, G.S. Pre-clinical evaluation of a themosensitive gel containing epothilone B and mTOR/Hsp90 targeted agents in an ovarian tumor model. J. Control. Release 2017, 268, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.T.; Chen, K.G.; Liu, A.; Huang, H.; Fan, Y.N.; Zhao, D.K.; Ye, Q.N.; Zhang, H.B.; Xu, C.F.; Shen, S.; et al. Immunomodulating nano-adaptors potentiate antibody-based cancer immunotherapy. Nat. Commun. 2021, 12, 1359. [Google Scholar] [CrossRef] [PubMed]
- Hoogstad-van Evert, J.S.; Maas, R.J.; van der Meer, J.; Cany, J.; van der Steen, S.; Jansen, J.H.; Miller, J.S.; Bekkers, R.; Hobo, W.; Massuger, L.; et al. Peritoneal NK cells are responsive to IL-15 and percentages are correlated with outcome in advanced ovarian cancer patients. Oncotarget 2018, 9, 34810–34820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nersesian, S.; Glazebrook, H.; Toulany, J.; Grantham, S.R.; Boudreau, J.E. Naturally Killing the Silent Killer: NK Cell-Based Immunotherapy for Ovarian Cancer. Front. Immunol. 2019, 10, 1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogstad-van Evert, J.S.; Bekkers, R.; Ottevanger, N.; Jansen, J.H.; Massuger, L.; Dolstra, H. Harnessing natural killer cells for the treatment of ovarian cancer. Gynecol. Oncol. 2020, 157, 810–816. [Google Scholar] [CrossRef]
- Sun, Y.; Yao, Z.; Zhao, Z.; Xiao, H.; Xia, M.; Zhu, X.; Jiang, X.; Sun, C. Natural killer cells inhibit metastasis of ovarian carcinoma cells and show therapeutic effects in a murine model of ovarian cancer. Exp. Ther. Med. 2018, 16, 1071–1078. [Google Scholar] [CrossRef]
- Geller, M.A.; Knorr, D.A.; Hermanson, D.A.; Pribyl, L.; Bendzick, L.; McCullar, V.; Miller, J.S.; Kaufman, D.S. Intraperitoneal delivery of human natural killer cells for treatment of ovarian cancer in a mouse xenograft model. Cytotherapy 2013, 15, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells 2016, 34, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Zhang, Q.Y.; Miao, Y.H.; Kang, W.Y.; Tian, Z.G.; Xu, D.M.; Xiao, W.H.; Fang, F. Interleukin-33 activates and recruits natural killer cells to inhibit pulmonary metastatic cancer development. Int. J. Cancer 2020, 146, 1421–1434. [Google Scholar] [CrossRef]
- Deng, X.; Terunuma, H.; Nieda, M.; Xiao, W.; Nicol, A. Synergistic cytotoxicity of ex vivo expanded natural killer cells in combination with monoclonal antibody drugs against cancer cells. Int. Immunopharmacol. 2012, 14, 593–605. [Google Scholar] [CrossRef]
- Liem, N.T.; Van Phong, N.; Kien, N.T.; Anh, B.V.; Huyen, T.L.; Thao, C.T.; Tu, N.D.; Hiep, D.T.; Hoai Thu, D.T.; Nhung, H.T.M. Phase I Clinical Trial Using Autologous Ex Vivo Expanded NK Cells and Cytotoxic T Lymphocytes for Cancer Treatment in Vietnam. Int. J. Mol. Sci. 2019, 20, 3166. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, N.A.P.; DeGolier, K.; Haberthur, K.; Chinn, H.; Moyes, K.W.; Bouchlaka, M.N.; Walker, K.L.; Capitini, C.M.; Crane, C.A. An Uncoupling of Canonical Phenotypic Markers and Functional Potency of Ex Vivo-Expanded Natural Killer Cells. Front. Immunol. 2018, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Michen, S.; Frosch, J.; Fussel, M.; Schackert, G.; Momburg, F.; Temme, A. Artificial feeder cells expressing ligands for killer cell immunoglobulin-like receptors and CD94/NKG2A for expansion of functional primary natural killer cells with tolerance to self. Cytotherapy 2020, 22, 354–368. [Google Scholar] [CrossRef]
- Yao, X.; Matosevic, S. Chemokine networks modulating natural killer cell trafficking to solid tumors. Cytokine Growth Factor Rev. 2021, 59, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, J.A.A.; Felder, M.; Horibata, S.; Belisle, J.A.; Kapur, A.; Holden, H.; Petrie, S.; Migneault, M.; Rancourt, C.; Connor, J.P.; et al. MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol. Cancer 2010, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nervi, B.; Rettig, M.P.; Ritchey, J.K.; Wang, H.L.; Bauer, G.; Walker, J.; Bonyhadi, M.L.; Berenson, R.J.; Prior, J.L.; Piwnica-Worms, D.; et al. Factors affecting human T cell engraftment, trafficking, and associated xenogeneic graft-vs-host disease in NOD/SCID beta2mnull mice. Exp. Hematol. 2007, 35, 1823–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Gras Navarro, A.; Bjorklund, A.T.; Chekenya, M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front. Immunol. 2015, 6, 202. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, A.; Cerwenka, A. Natural killer cells and solid tumors. J. Innate Immun. 2011, 3, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef] [PubMed]
- Greppi, M.; Tabellini, G.; Patrizi, O.; Candiani, S.; Decensi, A.; Parolini, S.; Sivori, S.; Pesce, S.; Paleari, L.; Marcenaro, E. Strengthening the AntiTumor NK Cell Function for the Treatment of Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, C.E.; Werner, B.; Hacker, N.F.; Warton, K. The untapped potential of ascites in ovarian cancer research and treatment. Br. J. Cancer 2020, 123, 9–16. [Google Scholar] [CrossRef]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Cortes, A.K.N.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef]
- Thangaraj, J.L.; Ahn, S.Y.; Jung, S.H.; Vo, M.C.; Chu, T.H.; Thi Phan, M.T.; Kwon, M.; Lee, K.H.; Kim, M.; Song, G.Y.; et al. Expanded natural killer cells augment the antimyeloma effect of daratumumab, bortezomib, and dexamethasone in a mouse model. Cell. Mol. Immunol. 2021, 18, 1652–1661. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Li, Y.; Wu, Y.; Xie, S.; Ma, J.; Yue, J.; Lv, R.; Tian, Z.; Fang, F.; Xiao, W. Anti-Tumor Activity of Expanded PBMC-Derived NK Cells by Feeder-Free Protocol in Ovarian Cancer. Cancers 2021, 13, 5866. https://doi.org/10.3390/cancers13225866
Chen M, Li Y, Wu Y, Xie S, Ma J, Yue J, Lv R, Tian Z, Fang F, Xiao W. Anti-Tumor Activity of Expanded PBMC-Derived NK Cells by Feeder-Free Protocol in Ovarian Cancer. Cancers. 2021; 13(22):5866. https://doi.org/10.3390/cancers13225866
Chicago/Turabian StyleChen, Minhua, Yutong Li, Yu Wu, Siqi Xie, Jie Ma, Jingjing Yue, Rong Lv, Zhigang Tian, Fang Fang, and Weihua Xiao. 2021. "Anti-Tumor Activity of Expanded PBMC-Derived NK Cells by Feeder-Free Protocol in Ovarian Cancer" Cancers 13, no. 22: 5866. https://doi.org/10.3390/cancers13225866
APA StyleChen, M., Li, Y., Wu, Y., Xie, S., Ma, J., Yue, J., Lv, R., Tian, Z., Fang, F., & Xiao, W. (2021). Anti-Tumor Activity of Expanded PBMC-Derived NK Cells by Feeder-Free Protocol in Ovarian Cancer. Cancers, 13(22), 5866. https://doi.org/10.3390/cancers13225866