
Directing the Future Breakthroughs in Immunotherapy: The Importance of a Holistic Approach to the Tumour Microenvironment


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of Immunotherapy
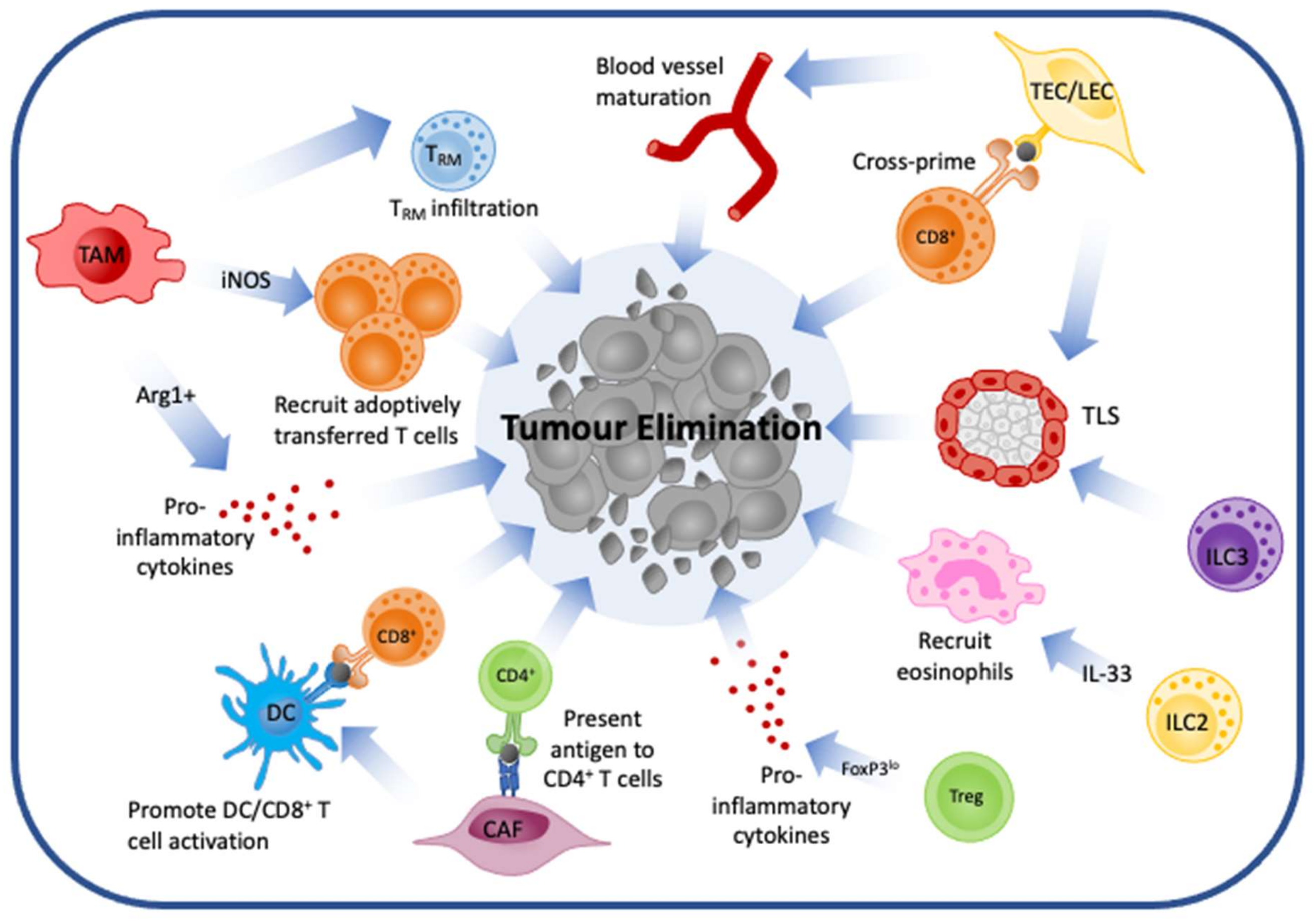
3. Cell Types
3.1. Cancer-Associated Fibroblasts
3.2. Tumour Endothelial Cells
3.3. Tumour-Associated Macrophages
3.4. Dendritic Cells
3.5. Myeloid-Derived Suppressor Cells
3.6. Innate Lymphoid Cells
3.7. Natural Killer Cells
3.8. B Cells
3.9. T Cells
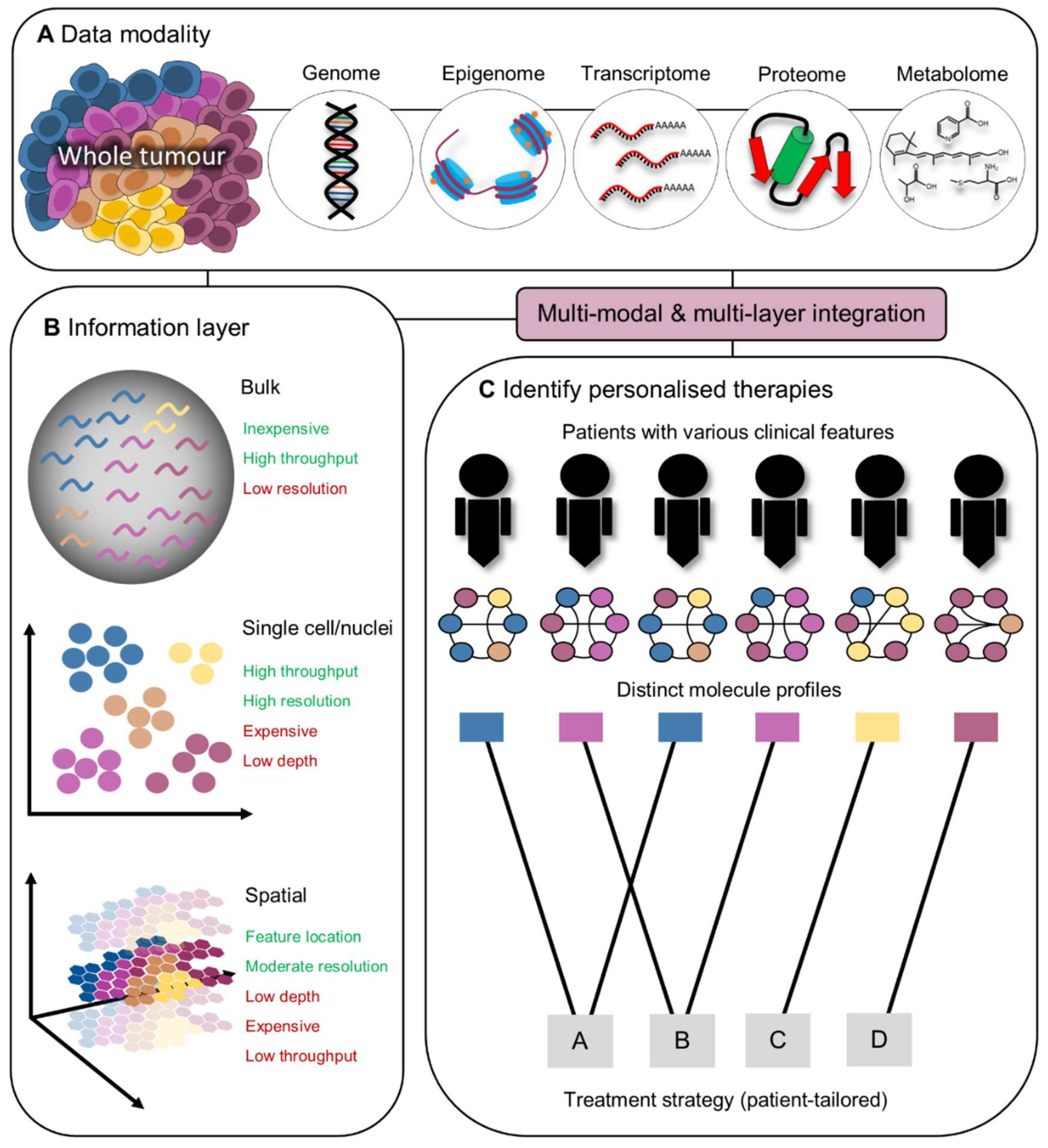
4. Development of Personalised Immunotherapies Guided by Integrated Omics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.D.; Zhao, H.; Wang, X.; Atkins, R.; Menges, M.; Reid, K.; Spitler, K.; Faramand, R.; Bachmeier, C.; Dean, E.A.; et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood 2021, 137, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Huff, A.L.; Wongthida, P.; Thompson, J.; Kottke, T.; Tonne, J.; Schuelke, M.; Ayasoufi, K.; Driscoll, C.B.; Shim, K.G.; et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020, 11, 3187. [Google Scholar] [CrossRef]
- Chen, G.M.; Chen, C.; Das, R.K.; Gao, P.; Chen, C.H.; Bandyopadhyay, S.; Ding, Y.Y.; Uzun, Y.; Yu, W.; Zhu, Q.; et al. Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discov. 2021, 11, 2186–2199. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharm. 2015, 23, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.A.; Dudley, M.E.; Rosenberg, S.A. Adoptive cell therapy: Genetic modification to redirect effector cell specificity. Cancer J. 2010, 16, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, N.A.; Nguyen, P.; Engle, E.L.; Kaunitz, G.J.; Cottrell, T.R.; Berry, S.; Green, B.; Soni, A.; Cuda, J.D.; Stein, J.E.; et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J. Immunother. Cancer 2018, 6, 99. [Google Scholar] [CrossRef]
- Stern, P.L. Harnessing immunity for therapy in human papillomavirus driven cancers. Tumour Virus Res. 2021, 11, 200212. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Lipson, E.J.; Tawbi, H.A.H.; Schadendorf, D.; Ascierto, P.A.; Matamala, L.; Gutierrez, E.C.; Rutkowski, P.; Gogas, H.; Lao, C.D.; Janoski de Menezes, J.; et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J. Clin. Oncol. 2021, 39, 9503. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: The molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 2020, 39, 112. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; Tang, Z.; Yin, S.; Mo, F.; Yang, X.; Hou, X.; Liu, A.; Lu, X. Fusion of Dendritic Cells and Cancer-Associated Fibroblasts for Activation of Anti-Tumor Cytotoxic T Lymphocytes. J. Biomed. Nanotechnol. 2018, 14, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwe, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99, 186–196. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironmen—New Findings and Future Perspectives. Front. Cell Dev. Biol. 2020, 8, 766. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Ugel, S.; Facciponte, J.; Facciabene, A. The dark side of tumor-associated endothelial cells. Semin. Immunol. 2018, 35, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.W.; Duraes, F.V.; Hirosue, S.; Raghavan, V.R.; Nembrini, C.; Thomas, S.N.; Issa, A.; Hugues, S.; Swartz, M.A. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012, 1, 191–199. [Google Scholar] [CrossRef]
- Vokali, E.; Yu, S.S.; Hirosue, S.; Rincon-Restrepo, M.; Fernanda, V.D.; Scherer, S.; Corthesy-Henrioud, P.; Kilarski, W.W.; Mondino, A.; Zehn, D.; et al. Lymphatic endothelial cells prime naive CD8(+) T cells into memory cells under steady-state conditions. Nat. Commun. 2020, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Goc, J.; Germain, C.; Vo-Bourgais, T.K.; Lupo, A.; Klein, C.; Knockaert, S.; de Chaisemartin, L.; Ouakrim, H.; Becht, E.; Alifano, M.; et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014, 74, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Posch, F.; Silina, K.; Leibl, S.; Mundlein, A.; Moch, H.; Siebenhuner, A.; Samaras, P.; Riedl, J.; Stotz, M.; Szkandera, J.; et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology 2018, 7, e1378844. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, N.; Ino, Y.; Yamazaki-Itoh, R.; Kanai, Y.; Kosuge, T.; Shimada, K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br. J. Cancer 2015, 112, 1782–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, L.; Le Guellec, S.; Filleron, T.; Lamant, L.; Meyer, N.; Rochaix, P.; Garrido, I.; Girard, J.P. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology 2012, 1, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Muraille, E.; Leo, O.; Moser, M. TH1/TH2 paradigm extended: Macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front. Immunol. 2014, 5, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef] [Green Version]
- Shimura, S.; Yang, G.; Ebara, S.; Wheeler, T.M.; Frolov, A.; Thompson, T.C. Reduced infiltration of tumor-associated macrophages in human prostate cancer: Association with cancer progression. Cancer Res. 2000, 60, 5857–5861. [Google Scholar] [PubMed]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Welsh, T.J.; Green, R.H.; Richardson, D.; Waller, D.A.; O’Byrne, K.J.; Bradding, P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 8959–8967. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e718. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Martin, E.M.; Mellows, T.W.P.; Clarke, J.; Ganesan, A.P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1(hot) tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef] [PubMed]
- Van Overmeire, E.; Stijlemans, B.; Heymann, F.; Keirsse, J.; Morias, Y.; Elkrim, Y.; Brys, L.; Abels, C.; Lahmar, Q.; Ergen, C.; et al. M-CSF and GM-CSF Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the Tumor Microenvironment. Cancer Res. 2016, 76, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef] [PubMed]
- Vogelpoel, L.T.; Hansen, I.S.; Rispens, T.; Muller, F.J.; van Capel, T.M.; Turina, M.C.; Vos, J.B.; Baeten, D.L.; Kapsenberg, M.L.; de Jong, E.C.; et al. Fc gamma receptor-TLR cross-talk elicits pro-inflammatory cytokine production by human M2 macrophages. Nat. Commun. 2014, 5, 5444. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Sozio, F.; Barbazza, I.; Salvi, V.; Tiberio, L.; Laffranchi, M.; Gismondi, A.; Bosisio, D.; Schioppa, T.; Sozzani, S. Functional Role of Dendritic Cell Subsets in Cancer Progression and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 3930. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. A new paradigm for tumor immune escape: Beta-catenin-driven immune exclusion. J. Immunother. Cancer 2015, 3, 43. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.; Diao, J.; Gu, H.; Khatri, I.; Zhao, J.; Cattral, M.S. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep. 2015, 13, 2851–2864. [Google Scholar] [CrossRef] [Green Version]
- Caronni, N.; Simoncello, F.; Stafetta, F.; Guarnaccia, C.; Ruiz-Moreno, J.S.; Opitz, B.; Galli, T.; Proux-Gillardeaux, V.; Benvenuti, F. Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer. Cancer Res. 2018, 78, 1685–1699. [Google Scholar] [CrossRef] [Green Version]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, R.; Tyurin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; Celis, E.; et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Audsley, K.M.; McDonnell, A.M.; Waithman, J. Cross-Presenting XCR1(+) Dendritic Cells as Targets for Cancer Immunotherapy. Cells 2020, 9, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e717. [Google Scholar] [CrossRef] [Green Version]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056.e1039. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.D.; Newnes, H.V.; McDonnell, A.; Bosco, A.; Waithman, J. Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression. Cells 2021, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Sinha, P.; Chornoguz, O.; Clements, V.K.; Artemenko, K.A.; Zubarev, R.A.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 2011, 117, 5381–5390. [Google Scholar] [CrossRef] [PubMed]
- Baniyash, M. Myeloid-derived suppressor cells as intruders and targets: Clinical implications in cancer therapy. Cancer Immunol. Immunother. 2016, 65, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [Green Version]
- Pico de Coana, Y.; Poschke, I.; Gentilcore, G.; Mao, Y.; Nystrom, M.; Hansson, J.; Masucci, G.V.; Kiessling, R. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol. Res. 2013, 1, 158–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef] [Green Version]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Cheng, F.; Yu, B.; Nefedova, Y.; Sotomayor, E.; Lush, R.; Gabrilovich, D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003, 63, 4441–4449. [Google Scholar]
- Weiss, J.M.; Subleski, J.J.; Back, T.; Chen, X.; Watkins, S.K.; Yagita, H.; Sayers, T.J.; Murphy, W.J.; Wiltrout, R.H. Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/alphaCD40 therapy. J. Immunol. 2014, 192, 5821–5829. [Google Scholar] [CrossRef] [Green Version]
- Tobin, R.P.; Jordan, K.R.; Robinson, W.A.; Davis, D.; Borges, V.F.; Gonzalez, R.; Lewis, K.D.; McCarter, M.D. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol 2018, 63, 282–291. [Google Scholar] [CrossRef]
- Melief, C.J.M.; Welters, M.J.P.; Vergote, I.; Kroep, J.R.; Kenter, G.G.; Ottevanger, P.B.; Tjalma, W.A.A.; Denys, H.; van Poelgeest, M.I.E.; Nijman, H.W.; et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci. Transl. Med. 2020, 12, 535. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seliger, B.; Ruiz-Cabello, F.; Garrido, F. IFN inducibility of major histocompatibility antigens in tumors. Adv. Cancer Res. 2008, 101, 249–276. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-gamma in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [Green Version]
- Messing, M.; Jan-Abu, S.C.; McNagny, K. Group 2 Innate Lymphoid Cells: Central Players in a Recurring Theme of Repair and Regeneration. Int. J. Mol. Sci. 2020, 21, 1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchard, M.; Ghiringhelli, F. ILC2s in cancer: Context matters. Nat. Immunol. 2021, 22, 804–806. [Google Scholar] [CrossRef]
- Ikutani, M.; Yanagibashi, T.; Ogasawara, M.; Tsuneyama, K.; Yamamoto, S.; Hattori, Y.; Kouro, T.; Itakura, A.; Nagai, Y.; Takaki, S.; et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J. Immunol. 2012, 188, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Jacquelot, N.; Seillet, C.; Wang, M.; Pizzolla, A.; Liao, Y.; Hediyeh-Zadeh, S.; Grisaru-Tal, S.; Louis, C.; Huang, Q.; Schreuder, J.; et al. Blockade of the co-inhibitory molecule PD-1 unleashes ILC2-dependent antitumor immunity in melanoma. Nat. Immunol. 2021, 22, 851–864. [Google Scholar] [CrossRef]
- Huang, Q.; Jacquelot, N.; Preaudet, A.; Hediyeh-Zadeh, S.; Souza-Fonseca-Guimaraes, F.; McKenzie, A.N.J.; Hansbro, P.M.; Davis, M.J.; Mielke, L.A.; Putoczki, T.L.; et al. Type 2 Innate Lymphoid Cells Protect against Colorectal Cancer Progression and Predict Improved Patient Survival. Cancers 2021, 13, 559. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Loiacono, F.; Di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, A.; Ogino, T.; Kayama, H.; Okuzaki, D.; Nishimura, J.; Fujino, S.; Miyoshi, N.; Takahashi, H.; Uemura, M.; Matsuda, C.; et al. Human NKp44(+) Group 3 Innate Lymphoid Cells Associate with Tumor-Associated Tertiary Lymphoid Structures in Colorectal Cancer. Cancer Immunol. Res. 2020, 8, 724–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenring, M.; vom Berg, J.; Kristiansen, G.; Saller, E.; Becher, B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 2010, 11, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Riggan, L.; Shah, S.; O’Sullivan, T.E. Arrested development: Suppression of NK cell function in the tumor microenvironment. Clin. Transl. Immunol. 2021, 10, e1238. [Google Scholar] [CrossRef]
- Habif, G.; Crinier, A.; Andre, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef]
- Nersesian, S.; Schwartz, S.L.; Grantham, S.R.; MacLean, L.K.; Lee, S.N.; Pugh-Toole, M.; Boudreau, J.E. NK cell infiltration is associated with improved overall survival in solid cancers: A systematic review and meta-analysis. Transl. Oncol. 2021, 14, 100930. [Google Scholar] [CrossRef]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Dong, W.; Wu, X.; Ma, S.; Wang, Y.; Nalin, A.P.; Zhu, Z.; Zhang, J.; Benson, D.M.; He, K.; Caligiuri, M.A.; et al. The Mechanism of Anti-PD-L1 Antibody Efficacy against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov. 2019, 9, 1422–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.; Bendell, J.C.; Han, S.W.; Naidoo, J.; Lieu, C.; Carneiro, B.A.; Varga, E.; Li, X.; Kourtesis, P.; Abdullah, S.E.; et al. 1201P—Durvalumab + monalizumab, mFOLFOX6, and bevacizumab in patients (pts) with metastatic microsatellite-stable colorectal cancer (MSS-CRC). Ann. Oncol. 2019, 30, v490–v491. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Grover, A.C.; Donald, E.J.; Carr, A.; Yu, J.; Whitfield, J.; Nelson, M.; Takeshita, N.; Chang, A.E. Simultaneous targeting of CD3 on T cells and CD40 on B or dendritic cells augments the antitumor reactivity of tumor-primed lymph node cells. J. Immunol. 2005, 175, 1424–1432. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.Y.; Gao, Q.; Wang, Z.C.; Zhou, J.; Wang, X.Y.; Min, Z.H.; Shi, Y.H.; Shi, G.M.; Ding, Z.B.; Ke, A.W.; et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5994–6005. [Google Scholar] [CrossRef] [Green Version]
- Carmi, Y.; Spitzer, M.H.; Linde, I.L.; Burt, B.M.; Prestwood, T.R.; Perlman, N.; Davidson, M.G.; Kenkel, J.A.; Segal, E.; Pusapati, G.V.; et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 2015, 521, 99–104. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Chen, J.; Tan, Y.; Sun, F.; Hou, L.; Zhang, C.; Ge, T.; Yu, H.; Wu, C.; Zhu, Y.; Duan, L.; et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020, 21, 152. [Google Scholar] [CrossRef]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, D.T.; Gabriel, S.S.; Chisanga, D.; Gloury, R.; Gubser, P.M.; Vasanthakumar, A.; Shi, W.; Kallies, A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 2020, 21, 1256–1266. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gurusamy, D.; et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef]
- Brummelman, J.; Mazza, E.M.C.; Alvisi, G.; Colombo, F.S.; Grilli, A.; Mikulak, J.; Mavilio, D.; Alloisio, M.; Ferrari, F.; Lopci, E.; et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J. Exp. Med. 2018, 215, 2520–2535. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpoux, A.; Lai, C.Y.; Hedrick, S.M.; Doedens, A.L. FOXO1 opposition of CD8(+) T cell effector programming confers early memory properties and phenotypic diversity. Proc. Natl. Acad. Sci. USA 2017, 114, E8865–E8874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speiser, D.E.; Utzschneider, D.T.; Oberle, S.G.; Munz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The Emerging Role of CD8(+) Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front. Immunol. 2018, 9, 1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, J.R.; Milne, K.; Watson, P.; Deleeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wu, S.; Zeng, H.; Liu, Z.; Dong, W.; He, W.; Chen, X.; Dong, X.; Zheng, L.; Lin, T.; et al. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J. Urol. 2015, 194, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Komdeur, F.L.; Prins, T.M.; van de Wall, S.; Plat, A.; Wisman, G.B.A.; Hollema, H.; Daemen, T.; Church, D.N.; de Bruyn, M.; Nijman, H.W. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology 2017, 6, e1338230. [Google Scholar] [CrossRef]
- Franciszkiewicz, K.; Le Floc’h, A.; Jalil, A.; Vigant, F.; Robert, T.; Vergnon, I.; Mackiewicz, A.; Benihoud, K.; Validire, P.; Chouaib, S.; et al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. 2009, 69, 6249–6255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Blanc, C.; Hans, S.; Tran, T.; Granier, C.; Saldman, A.; Anson, M.; Oudard, S.; Tartour, E. Targeting Resident Memory T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1722. [Google Scholar] [CrossRef] [Green Version]
- Brentville, V.A.; Metheringham, R.L.; Daniels, I.; Atabani, S.; Symonds, P.; Cook, K.W.; Vankemmelbeke, M.; Choudhury, R.; Vaghela, P.; Gijon, M.; et al. Combination vaccine based on citrullinated vimentin and enolase peptides induces potent CD4-mediated anti-tumor responses. J. Immunother. Cancer 2020, 8, e000560. [Google Scholar] [CrossRef] [PubMed]
- Hoepner, S.; Loh, J.M.; Riccadonna, C.; Derouazi, M.; Maroun, C.Y.; Dietrich, P.Y.; Walker, P.R. Synergy between CD8 T cells and Th1 or Th2 polarised CD4 T cells for adoptive immunotherapy of brain tumours. PLoS ONE 2013, 8, e63933. [Google Scholar] [CrossRef]
- Goldstein, M.J.; Kohrt, H.E.; Houot, R.; Varghese, B.; Lin, J.T.; Swanson, E.; Levy, R. Adoptive cell therapy for lymphoma with CD4 T cells depleted of CD137-expressing regulatory T cells. Cancer Res. 2012, 72, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Haabeth, O.A.; Tveita, A.; Fauskanger, M.; Hennig, K.; Hofgaard, P.O.; Bogen, B. Idiotype-specific CD4(+) T cells eradicate disseminated myeloma. Leukemia 2016, 30, 1216–1220. [Google Scholar] [CrossRef]
- Hong, S.; Qian, J.; Yang, J.; Li, H.; Kwak, L.W.; Yi, Q. Roles of idiotype-specific t cells in myeloma cell growth and survival: Th1 and CTL cells are tumoricidal while Th2 cells promote tumor growth. Cancer Res. 2008, 68, 8456–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Hunder, N.N.; Wallen, H.; Cao, J.; Hendricks, D.W.; Reilly, J.Z.; Rodmyre, R.; Jungbluth, A.; Gnjatic, S.; Thompson, J.A.; Yee, C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med. 2008, 358, 2698–2703. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Han, N.I.; Yoon, S.K. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 2019, 9, 3260. [Google Scholar] [CrossRef] [Green Version]
- Mattes, J.; Hulett, M.; Xie, W.; Hogan, S.; Rothenberg, M.E.; Foster, P.; Parish, C. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: An eotaxin and STAT6-dependent process. J. Exp. Med. 2003, 197, 387–393. [Google Scholar] [CrossRef]
- Shen, Y.; Fujimoto, S. A tumor-specific Th2 clone initiating tumor rejection via primed CD8+ cytotoxic T-lymphocyte activation in mice. Cancer Res. 1996, 56, 5005–5011. [Google Scholar]
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627. [Google Scholar] [CrossRef]
- Lorvik, K.B.; Hammarstrom, C.; Fauskanger, M.; Haabeth, O.A.; Zangani, M.; Haraldsen, G.; Bogen, B.; Corthay, A. Adoptive Transfer of Tumor-Specific Th2 Cells Eradicates Tumors by Triggering an In Situ Inflammatory Immune Response. Cancer Res. 2016, 76, 6864–6876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, P.A.; Klawon, D.E.J.; Miller, C.H. Regulatory T Cell Development. Annu. Rev. Immunol. 2020, 38, 421–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabi, A.; Tsokos, M.G.; Ding, Y.; Malek, T.R.; Klatzmann, D.; Tsokos, G.C. Regulatory T cells in the treatment of disease. Nat. Rev. Drug Discov. 2018, 17, 823–844. [Google Scholar] [CrossRef]
- Owen, D.L.; Sjaastad, L.E.; Farrar, M.A. Regulatory T Cell Development in the Thymus. J. Immunol. 2019, 203, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guldner, I.H.; Golomb, S.M.; Sun, L.; Harris, J.A.; Lu, X.; Zhang, S. Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nat. Commun. 2019, 10, 3817. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, K.; Shirai, M.; Taniguchi, K.; Hosoi, A.; Sun, C.; Kobayashi, Y.; Maejima, K.; Fujita, M.; Nakagawa, H.; Nomura, S.; et al. Deep immunophenotyping at the single-cell level identifies a combination of anti-IL-17 and checkpoint blockade as an effective treatment in a preclinical model of data-guided personalized immunotherapy. J. Immunother. Cancer 2020, 8, e001358. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- De Jong, E.; Bosco, A. Unlocking immune-mediated disease mechanisms with transcriptomics. Biochem. Soc. Trans. 2021, 49, 705–714. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Smith, S.E.; Chen, S.; Li, H.; Wu, D.; Meneses-Giles, P.I.; Wang, Y.; Hembree, M.; Yi, K.; Zhao, X.; et al. Tumor-initiating stem cell shapes its microenvironment into an immunosuppressive barrier and pro-tumorigenic niche. Cell Rep. 2021, 36, 109674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e429. [Google Scholar] [CrossRef]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021, 39, 649–661.e645. [Google Scholar] [CrossRef] [PubMed]
- Vathiotis, I.A.; Yang, Z.; Reeves, J.; Toki, M.; Aung, T.N.; Wong, P.F.; Kluger, H.; Syrigos, K.N.; Warren, S.; Rimm, D.L. Models that combine transcriptomic with spatial protein information exceed the predictive value for either single modality. NPJ Precis. Oncol. 2021, 5, 45. [Google Scholar] [CrossRef]
- Gouin, K.H., 3rd; Ing, N.; Plummer, J.T.; Rosser, C.J.; Ben Cheikh, B.; Oh, C.; Chen, S.S.; Chan, K.S.; Furuya, H.; Tourtellotte, W.G.; et al. An N-Cadherin 2 expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer. Nat. Commun. 2021, 12, 4906. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Grauel, A.L.; Nguyen, B.; Ruddy, D.; Laszewski, T.; Schwartz, S.; Chang, J.; Chen, J.; Piquet, M.; Pelletier, M.; Yan, Z.; et al. TGFbeta-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. Commun. 2020, 11, 6315. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Denti, V.; Mahajneh, A.; Capitoli, G.; Clerici, F.; Piga, I.; Pagani, L.; Chinello, C.; Bolognesi, M.M.; Paglia, G.; Galimberti, S.; et al. Lipidomic Typing of Colorectal Cancer Tissue Containing Tumour-Infiltrating Lymphocytes by MALDI Mass Spectrometry Imaging. Metabolites 2021, 11, 599. [Google Scholar] [CrossRef]
- Berghmans, E.; Jacobs, J.; Deben, C.; Hermans, C.; Broeckx, G.; Smits, E.; Maes, E.; Raskin, J.; Pauwels, P.; Baggerman, G. Mass Spectrometry Imaging Reveals Neutrophil Defensins as Additional Biomarkers for Anti-PD-(L)1 Immunotherapy Response in NSCLC Patients. Cancers 2020, 12, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Xia, F.; Shukla, M.; Brettin, T.; Garcia-Cardona, C.; Cohn, J.; Allen, J.E.; Maslov, S.; Holbeck, S.L.; Doroshow, J.H.; Evrard, Y.A.; et al. Predicting tumor cell line response to drug pairs with deep learning. BMC Bioinform. 2018, 19, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newnes, H.V.; Armitage, J.D.; Audsley, K.M.; Bosco, A.; Waithman, J. Directing the Future Breakthroughs in Immunotherapy: The Importance of a Holistic Approach to the Tumour Microenvironment. Cancers 2021, 13, 5911. https://doi.org/10.3390/cancers13235911
Newnes HV, Armitage JD, Audsley KM, Bosco A, Waithman J. Directing the Future Breakthroughs in Immunotherapy: The Importance of a Holistic Approach to the Tumour Microenvironment. Cancers. 2021; 13(23):5911. https://doi.org/10.3390/cancers13235911
Chicago/Turabian StyleNewnes, Hannah V., Jesse D. Armitage, Katherine M. Audsley, Anthony Bosco, and Jason Waithman. 2021. "Directing the Future Breakthroughs in Immunotherapy: The Importance of a Holistic Approach to the Tumour Microenvironment" Cancers 13, no. 23: 5911. https://doi.org/10.3390/cancers13235911
APA StyleNewnes, H. V., Armitage, J. D., Audsley, K. M., Bosco, A., & Waithman, J. (2021). Directing the Future Breakthroughs in Immunotherapy: The Importance of a Holistic Approach to the Tumour Microenvironment. Cancers, 13(23), 5911. https://doi.org/10.3390/cancers13235911