
Propensity for Early Metastatic Spread in Breast Cancer: Role of Tumor Vascularization Features and Tumor Immune Infiltrate


Abstract
:Simple Summary
Abstract
1. Introduction
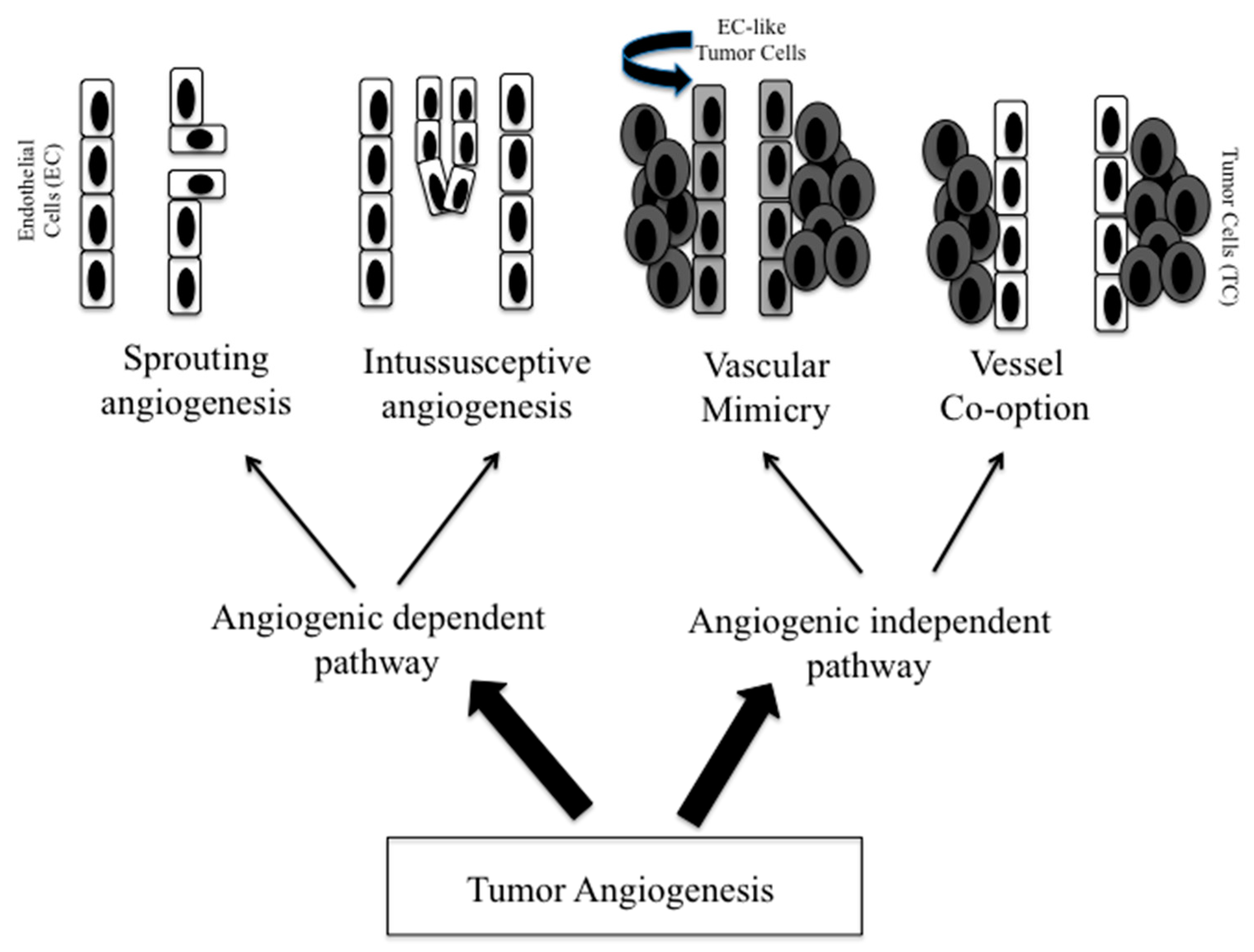
2. Tumor Vasculature
2.1. Sprouting and Intussusceptive Angiogenesis
2.1.1. Molecular Mechanisms of Sprouting and Intussusceptive Angiogenesis
2.1.2. Immunohistochemical Assessment of Sprouting and Intussusceptive Angiogenesis
2.2. Vascular Mimicry
2.3. Vessel Co-Option
3. Metastasis Propensity and Tumor Microenvironment (TME)
3.1. Molecular Mechanisms of the Influence of TME on Breast Cancer Metastasis
3.1.1. EMT and TME
3.1.2. Angiogenesis and TME
3.1.3. ECM Remodeling and TME
3.1.4. Intravasation/Survival in Circulation and TME
3.2. Tumor-Microenvironment Components
3.2.1. Tumor-Associated Macrophages (TAMs)
3.2.2. Tumor-Infiltrating Lymphocytes (TILs)
3.2.3. Neutrophils
3.2.4. Cancer-Associated Fibroblasts (CAFs)
3.2.5. Cancer-Associated Adipocytes (CAAs)
3.2.6. Cytokines
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Madelblatt, J.S.; Kurian, A.W. Change in survival in metastatic breast cancer with treatment advances: Meta-analysis and systematic review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, O.J.; Bay, B.-H.; Yip, G.; Yu, Y. Breast cancer metastasis. Cancer Genom. Proteom. 2012, 9, 311–320. [Google Scholar]
- Deryugina, E.I.; Kiosses, W.B. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017, 19, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narod, S.A.; Sopik, V. Is invasion a necessary step for metastases in breast cancer? Breast Cancer Res. Treat. 2018, 169, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narod, S.A.; Iqbal, J.; Giannakeas, V.; Sopik, V.; Sun, P. Breast Cancer Mortality After a Diagnosis of Ductal Carcinoma In Situ. JAMA Oncol. 2015, 1, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Elshof, L.E.; Schmidt, M.; Rutgers, E.J.; van Leeuwen, F.E.; Wesseling, J.; Schaapveld, M. Cause-specific Mortality in a Population-based Cohort of 9799 Women Treated for Ductal Carcinoma in Situ. Ann. Surg. 2018, 267, 952–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadsten, C.; Garmo, H.; Fredriksson, I.; Sund, M.; Wärnberg, F. Risk of death from breast cancer after treatment for ductal carcinoma in situ. BJS 2017, 104, 1506–1513. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor Angiogenesis and Metastasis—Correlation in Invasive Breast Carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bluff, J.E.; Menakuru, S.R.; Cross, S.S.; Higham, S.E.; Balasubramanian, S.P.; Brown, N.J.; Reed, M.W.; Staton, C.A. Angiogenesis is associated with the onset of hyperplasia in human ductal breast dis-ease. Br. J. Cancer 2009, 101, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, P.M.; Chen, W.-P.; Mendez, A.; McLaren, C.E.; Su, M.-Y. Angiogenesis in the Progression of Breast Ductal Proliferations. Int. J. Surg. Pathol. 2009, 19, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Teo, N.B.; Shoker, B.S.; Jarvis, C.; Martin, L.; Sloane, J.P.; Holcombe, C. Vascular density and phenotype around ductal carcinoma in situ (DCIS) of the breast. Br. J. Cancer 2002, 86, 905–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna Priya, S.; Nagare, R.P.; Sneha, V.S.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T.S. Tumor angiogenesis-origin of blood vessels. Int J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Rohan, T.E.; Xue, X.; Lin, H.-M.; D’Alfonso, T.M.; Ginter, P.S.; Oktay, M.H.; Robinson, B.D.; Ginsberg, M.; Gertler, F.B.; Glass, A.G.; et al. Tumor Microenvironment of Metastasis and Risk of Distant Metastasis of Breast Cancer. J. Natl. Cancer Inst. 2014, 106, dju136. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.E.; Harney, A.S.; Pollard, J.W. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell 2016, 30, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, T.T.; Prechtl, A.M.; Pearson, G.W. Breast cancer subtype-specific interactions with the microenvironment dictate mecha-nisms of invasion. Cancer Res. 2011, 71, 6857–6866. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Re-quired for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annaratone, L.; Cascardi, E.; Vissio, E.; Sarotto, I.; Chmielik, E.; Sapino, A.; Berrino, E.; Marchiò, C. The multifaceted nature of tumor microenvironment in breast cancer. Pathobiology 2020, 87, 125–142. [Google Scholar] [CrossRef] [PubMed]
- North, S.; Moenner, M.; Bikfalvi, A. Recent developments in the regulation of the angiogenic swithc by cellular stress factors in tumors. Cancer Lett. 2005, 218, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.; Alves, T.; Fang, J. FGF-Dependent Metabolic Control of Vascular Development. J. Vasc. Surg. 2017, 66, 959. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.-P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1alpha in endothelial cells disrupts a hypoxiadriven VEGF autocrine loop neces-sary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.W.; Lewis, M.M.; Lawson, D.; Yin-Goen, Q.; Birsong, G.G.; Cotsonis, G.A.; Cohen, C.; Young, A.N. Angiogenic and lymphangiogenic microvessel density in breast carcinoma: Correlation with clinicopathologic parameters and VEGF-family gene expression. Mod. Pathol. 2005, 18, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.P.; Miller, K.D. Angiogenesis of breast cancer. J. Clin. Oncol. 2005, 23, 1782–1790. [Google Scholar] [CrossRef]
- Kim, J. Pericytes in Breast Cancer. Single Mol. Single Cell Seq. 2019, 1147, 93–107. [Google Scholar] [CrossRef]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Diéguez-Hurtado, R.; Schmidt, I.; Nakayama, M.; Jeong, H.-W.; Arf, H.; Adams, S.; Ferrara, N.; Adams, R.H. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, K.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. MCR 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2013, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, U.; Scharpfenecker, M.; Koidl, S.; Hegen, A.; Grunow, V.; Schmidt, J.M.; Kriz, W.; Thurston, G.; Augustin, H.G. The Tie-2 ligand Angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 2004, 103, 4150–4156. [Google Scholar] [CrossRef]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte Depletion Results in Hypoxia-Associated Epithelial-to-Mesenchymal Transition and Metastasis Mediated by Met Signaling Pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [Green Version]
- Keskin, D.; Kim, J.; Cooke, V.G.; Wu, C.-C.; Sugimoto, H.; Gu, C.; De Palma, M.; Kalluri, R.; LeBleu, V.S. Targeting Vascular Pericytes in Hypoxic Tumors Increases Lung Metastasis via Angiopoietin-2. Cell Rep. 2015, 10, 1066–1081. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Schrenk, S.; Goines, J.; Davis, G.E.; Boscolo, E. Constitutive Active Mutant TIE2 Induces Enlarged Vascular Lumen Formation with Loss of Apico-basal Polarity and Pericyte Recruitment. Sci. Rep. 2019, 9, 12352-12. [Google Scholar] [CrossRef]
- Venneri, M.A.; De Palma, M.; Ponzoni, M.; Pucci, F.; Scielzo, C.; Zonari, E.; Mazzieri, R.; Doglioni, C.; Naldini, L. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 2007, 109, 5276–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teichert, M.; Milde, L.; Holm, A.; Stanicek, L.; Gengenbacher, N.; Savant, S.; Ruckdeschel, T.; Hasanov, Z.; Srivastava, K.; Hu, J.; et al. Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nat. Commun. 2017, 8, 16106. [Google Scholar] [CrossRef] [PubMed]
- Burri, P.H.; Djonov, V. Intussusceptive angiogenesis--the alternative to capillary sprouting. Mol. Aspects Med. 2002, 23, S1–S27. [Google Scholar] [CrossRef]
- Hlushchuk, R.; Riesterer, O.; Baum, O.; Wood, J.; Gruber, G.; Pruschy, M.; Djonov, V. Tumor recovery by angiogenic switch from sprouting to intussusceptive angiogenesis after treatment with PTK787/ZK222584 or ionizing radiation. Am. J. Pathol. 2008, 173, 1173–1185. [Google Scholar] [CrossRef] [Green Version]
- Uzzan, B.; Nicolas, P.; Cucherat, M.; Perret, G.Y. Microvessel Density as a Prognostic Factor in Women with Breast Cancer: A Sys-tematic Review of the Literature and Meta-Analysis. Cancer Res. 2004, 64, 2941–2955. [Google Scholar] [CrossRef] [Green Version]
- Shrivastav, S.; Bal, A.; Singh, G.; Joshi, K. Tumor angiogenesis in breast cancer: Pericytes and maturation does not correlate with lymphnode metastasis and molecular subtypes. Clin. Breast Cancer 2016, 16, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Boulos, F.; Jabbour, M.; Mahfoud, Z.; Fakhruddin, N.; El-Sabban, M. Expression of HIF-1α and Markers of Angiogenesis Are Not Significantly Different in Triple Negative Breast Cancer Compared to Other Breast Cancer Molecular Subtypes: Implications for Future Therapy. PLoS ONE 2015, 10, e0129356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, A.; Holzer, T.; Chen, M.; Man, M.Z.; Schade, A.E. Differential expression of VEGFR2 protein in HER2 positive primary human breast cancer: Potential relevance to anti-angiogenic therapies. Cancer Cell Int. 2017, 17, 56. [Google Scholar] [CrossRef]
- Kraby, M.R.; Opdahl, S.; Akslen, L.A.; Bofin, A.M. Quantifying tumour vascularity in non-luminal breast cancers. J. Clin. Pathol. 2017, 70, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Kraby, M.R.; Krüger, K.; Opdahl, S.; Vatten, L.J.; Akslen, L.A.; Bofin, A.M. Microvascular proliferation in luminal A and basal-like breast cancer subtypes. J. Clin. Pathol. 2015, 68, 891–897. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; MacKay, D.; Pollard, J.W.; Lewis, C.E. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [Green Version]
- Gibby, K.; You, W.K.; Kadoya, K.; Helgadottir, H.; Young, L.J.; Ellies, L.G.; Chang, Y.; Cardiff, R.D.; Stallcup, W.B. Early vascular deficits are correlated with delayed mammary tumorigenesis in the MMTV-PyMT transgenic mouse following genetic ablation of the NG2 proteoglycan. Breast Cancer Res. 2012, 14, R67. [Google Scholar] [CrossRef] [Green Version]
- Paulsson, J.; Sjöblom, T.; Micke, P.; Pontén, F.; Landberg, G.; Heldin, C.-H.; Bergh, J.; Brennan, D.J.; Jirström, K.; Östman, A. Prognostic Significance of Stromal Platelet-Derived Growth Factor β-Receptor Expression in Human Breast Cancer. Am. J. Pathol. 2009, 175, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; de Sampaio, P.C.; Lundy, D.M.; Peng, Q.; Evans, K.W.; Sugimoto, H.; Gagea, M.; Kienast, Y.; do Amaral, N.S.; Rocha, R.M.; et al. Heterogeneous perivascular cell coverage affects breast cancer metastasis and re-sponse to chemotherapy. JCI Insight. 2016, 1, e90733. [Google Scholar] [CrossRef] [Green Version]
- Jansson, S.; Aaltonen, K.; Bendhal, P.O.; Falck, A.-K.; Karlsson, M.; Pietras, K.; Ryden, L. The PDGF pathway in breast cancer is linked to tumor aggressiveness, triple-negative subtype and early recurrence. Breast Cancer Res. Treat. 2018, 169, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Bao, M.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: A systemic review and meta-analysis. Eur. J. Cancer 2013, 49, 3914–3923. [Google Scholar] [CrossRef]
- Shen, Y.; Quan, J.; Wang, M.; Lixs, S.; Yang, J.; Lv, M.; Chen, Z.; Zhang, L.; Zhao, X.; Yang, J. Tumor vasculogenic mimicry formation as an unfavorable prognostic indicator in patients with breast cancer. Oncotarget 2017, 8, 56408–56416. [Google Scholar] [CrossRef]
- Hori, A.; Shimoda, M.; Naoi, Y.; Kagara, N.; Tanei, T.; Miyake, T.; Shimazu, K.; Kim, S.J.; Noguchi, S. Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Res. 2019, 21, 88. [Google Scholar] [CrossRef]
- Pezzella, F.; Di Bacco, A.; Andreola, S.; Nicholson, A.; Pastorino, U.; Harris, A. Angiogenesis in primary lung cancer and lung secondaries. Eur. J. Cancer 1996, 32, 2494–2500. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2016, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2016, 108, djw030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.R.; Wotherspoon, A.; Gao, Z.-H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adighibe, O.; Leek, R.D.; Fernandez-Mercado, M.; Hu, J.; Snell, C.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Why some tumours trigger neovascularisation and others don’t: The story thus far. Chin. J. Cancer 2016, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, C.L.T.; Forsare, C.; Bendhar, P.-O.; Falck, A.-K.; Ferno, M.; Lovgren, K.; Aaltonen, K.; Ryden, L. Expression of epithelial-mesenchymal transition-related markers and pheno-types during brest cancer progression. Breast Cancer Res. Treat. 2020, 181, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Kwang, W.; Zhou, Q.; Zhang, Y. TGF.B1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-B-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with spe-cial emphasis on the role of matrix metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012, 4, a011908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, Y.; Yu, W.; Zheng, S.; Zhang, S.; Sun, L.; Ding, K. Small role with big impact: miRNAs as communicators in the cross-talk between cancer-associated fibroblasts and cancer cells. Int. J. Biol. Sci. 2017, 13, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Wang, Y.; Yuan, Z.; Wang, S.; Du, H.; Liu, X.; Wang, Q.; Zhu, X. Human adipose-derived mesenchymal stem cells promote breast cancer MCF7 cell epithelial-mesenchymal transition by cross interacting with the TGF-β/Smad and PI3K/AKT signaling pathways. Mol. Med. Rep. 2019, 9, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.N.; Gao, H.; Anfossi, S.; Mego, M.; Reddy, N.G.; Debeb, B.; Giordano, A.; Tin, S.; Wu, Q.; Garza, R.J.; et al. Inflammation mediated metastasis: Immune induced epithelial-to-Mesenchymal transition in inflammatory breast Cancer cells. PLoS ONE 2015, 10, e0132710. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hy-poxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Chen, Y.S.; Yao, Y.D.; Chen, J.-Q.; Chen, J.-N.; Huang, S.-Y.; Zeng, Y.-J.; Yao, H.-R.; Zeng, S.-H.; Fu, Y.-S.; et al. CCL18 from tumour-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 2015, 6, 34758–34773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Li, J.-F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Quian, H.; Xue, X.-N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of brest cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.; Tal, A.O.; Scholz, A.; De Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 Regulates Gene Expression in TIE2-Expressing Monocytes and Augments Their Inherent Proangiogenic Functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watnick, R.S. The Role of the Tumor Microenvironment in Regulating Angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-Regulatory Cells: Key Players in Tumor Immune Escape and Angiogenesis: Figure 1. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angio-genesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wi, C.; Li, X. The potential role and staus of IL-17 family cytokines in breast cancer. Int. Immunoph. 2021, 95, 107544. [Google Scholar] [CrossRef]
- Guo, N.; Shen, G.; Zhang, Y.; Moustafa, A.A.; Ge, D.; You, Z. IL-17 promotes migration and invasion of human cancer cells through up-regulation of MTA1 expression. Front. Oncol. 2019, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenesis switch in a mouse model of multi-stage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, I.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and im-mune cell infiltration. Integr. Biol. (CAMB) 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Hou, Y.; Yang, G.; Wang, X.; Tang, S.; Du, Y.-E.; Yang, L.; Yu, T.; Zhang, H.; Zhou, M.; et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2015, 23, 132–145. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.-T.; Chi, J.-T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef]
- Stuelten, C.H.; DaCosta Byfield, S.; Arany, P.R.; Karpova, T.S.; Stetler.Stevenson, W.G.; Roberts, A.B. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNFa and TGFb. J. Cell Sci. 2005, 118, 2143–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangaletti, S.; DI Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-Derived SPARC Bridges Tumor Cell-Extracellular Matrix Interactions toward Metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Deng, C.-X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Khuon, S.; Liang, L.; Dettman, R.W.; Sporn, P.H.S.; Wysolmerski, R.B.; Chew, T.-L. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: A three-dimensional FRET study. J. Cell Sci. 2010, 123, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial–mesenchymal plasticity. Life Sci. Alliance 2019, 2, e201900425. [Google Scholar] [CrossRef] [PubMed]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.; Elbaz, M.; Cuitiño, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shani, O.; Raz, Y.; Monteran, L.; Scharff, Y.; Levi-Galibov, O.; Megides, O.; Shacham, H.; Cohen, N.; Silverbush, D.; Avivi, C.; et al. Evolution of metastases-associated fibroblasts in the lung microenvironment is driven by stage-specific transcriptional plasticity. eLife 2021, 10, e60745. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumor cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Miklikova, S.; Minarik, G.; Sedlackova, T.; Plava, J.; Cihova, M.; Jurisova, S.; Kalavska, K.; Karaba, M.; Benca, J.; Smolkova, B.; et al. Inflammation-Based Scores Increase the Prognostic Value of Circulating Tumor Cells in Primary Breast Cancer. Cancers 2020, 12, 1134. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.-F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct Visualization of Macrophage-Assisted Tumor Cell Intravasation in Mammary Tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage–Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, B.D.; Sica, G.L.; Liu, Y.-F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor Microenvironment of Metastasis in Human Breast Carcinoma: A Potential Prognostic Marker Linked to Hematogenous Dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparano, J.A.; Gray, R.; Oktay, M.H.; Entenberg, D.; Rohan, T.; Xue, X.; Donovan, M.; Peterson, M.; Shuber, A.; Hamilton, D.A.; et al. A metastasis biomarker (MetaSite Breast™ Score) is associated with distant recurrence in hormone receptor-positive, HER2-negative early-stage breast cancer. NPJ Breast Cancer 2017, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.S.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Torres, I.R.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. International TILs Working Group 2014. The evaluation of tumor-infiltrating lym-phocytes(TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Savas, P.; Luen, S.J.; Virassamy, B.; Salgado, R.; Loi, S. Clinical Validity and Utility of Tumor-Infiltrating Lymphocytes in Routine Clinical Practice for Breast Cancer Patients: Current and Future Directions. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamy, A.-S.; Bonsang-Kitzis, H.; De Croze, D.; Laas, E.; Darrigues, L.; Topciu, L.; Menet, E.; Vincent-Salomon, A.; Lerebours, F.; Pierga, J.-Y.; et al. Interaction between Molecular Subtypes and Stromal Immune Infiltration before and after Treatment in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Clin. Cancer Res. 2019, 25, 6731–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkert, C.; von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.-L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predic-tive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic re-view and meta-analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef] [Green Version]
- Metelli, A.; Wu, B.X.; Fugle, C.W.; Rachidi, S.; Sun, S.; Zhang, Y.; Wu, J.; Tomlinson, S.; Howe, P.H.; Yang, Y.; et al. Surface Expression of TGFβ Docking Receptor GARP Promotes Oncogenesis and Immune Tolerance in Breast Cancer. Cancer Res. 2016, 76, 7106–7117. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yu, P.F.; Han, Y.Y.; Lin, L.Y.; Sun, W.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. TNFa-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar]
- Iwase, T.; Sangai, T.; Sakakibara, M.; Sakakibara, J.; Ishigami, E.; Hayama, S.; Nakagawa, A.; Masuda, T.; Tabe, S.; Nagashima, T. An increased neutrophil-to-lymphocyte ratio predicts poorer survival following recur-rence for patients with breast cancer. Mol. Clin. Oncol. 2017, 6, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.S.; Sarnella, A.; D’Avino, G.; Zannetti, A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin. Cancer Biol. 2020, 60, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein-Achiasaf, L.; Morein, D.; Ben-Yaakov, H.; Liubomirski, Y.; Meshel, T.; Elbaz, E.; Dorot, O.; Pichinuk, E.; Gershovits, M.; Weil, M.; et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers 2021, 13, 1472. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1–TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2014, 34, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Xu, F.; Zhong, K.; Wang, S.; Huang, L.; Chen, W. Activated Tumor-infiltrating Fibroblasts Predict Worse Prognosis in Breast Cancer Patients. J. Cancer 2018, 9, 3736–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast can-cer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Fujisaki, K.; Fujimoto, H.; Sangai, T.; Nagashima, T.; Sakakibara, M.; Shiina, N.; Kuroda, M.; Aoyagi, Y.; Miyazaki, M. Cancer-mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res. Treat. 2015, 150, 255–263. [Google Scholar] [CrossRef]
- Wei, X.; Li, S.; He, J.; Du, H.; Liu, Y.; Yu, W.; Hu, H.; Han, L.; Wang, C.; Li, H.; et al. Tumor-secreted PAI-1 promotes breast cancer metastasis via the induction of adipocyte-derived colla-gen remodeling. Cell Commun. Signal. 2019, 17, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, R.; Kluz, P.; Tan, W.Z.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F.; et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopietin-like 4. Oncogene 2019, 38, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Attanè, C.; Milhas, D.; Hoy, A.; Muller, C. Metabolic remodeling induced by adipocytes: A new achilles’ heel in invasuve breast cancer? Curr. Med. Chem. 2020, 27, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Esquivel-Velazquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Monton, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, X.; Zhang, Q.; Cao, R.; Liu, J.; Zhong, J.; Wen, G.; Cao, D. Transforming growth factor-b signaling in tumor initiation, progression and therapy in breast cancer: An update. Cell Tissue Res. 2012, 347, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1–SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial–mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.E.; Schwertfeger, K.L. Proinflammatory cytokines in breast cancer: Mechanisms of action and potential targets for therapeutics. Curr. Drug Targets 2010, 11, 1133–1146. [Google Scholar] [CrossRef]
- Culig, Z. Cytokine disbalance in common human cancers. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Sasser, A.K.; Sullivan, N.J.; Studebaker, A.W.; Hendey, L.F.; Axel, A.E.; Hall, B.M. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007, 21, 3763–3770. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Kamel, M.; Shouman, S.; El-Merzebany, M.; Kilic, G.; Veenstra, T.; Saeed, M.; Wagih, M.; Diaz-Arrastia, C.; Patel, D.; Salama, S. Effect of Tumour Necrosis Factor-Alpha on Estrogen Metabolic Pathways in Breast Cancer Cells. J. Cancer 2012, 3, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Huang, R.; Chen, L.; Li, S.; Shi, Q.; Jordan, C.; Huang, R.-P. Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int. J. Cancer 2004, 109, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, X.; Hao, Y.; Li, J. Obesity-related protein biomarkers for predicting breast cancer risk: An overview of systematic reviews. Breast Cancer 2020, 28, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Caldefie-Chézet, F.; Damez, M.; de Latour, M.; Konska, G.; Mishellani, F.; Fusillier, C.; Guerry, M.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin: A proliferative factor for breast cancer? Study on human ductal car-cinoma. Biochem Biophys Res. Commun 2005, 334, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Jardé, T.; Caldefie-Chézet, F.; Damez, M.; Mishellany, F.; Penault.Llorca, F.; Guillot, J.; Vasson, M.P. Leptin and leptin receptor involvement in cancer development: A study on hu-man primary breast carcinoma. Oncol Rep. 2008, 19, 905–911. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Mean Differences/ (95% CI) | p | Hazard Ratio (95% CI) | p | |
|---|---|---|---|---|
| MVD, microvessels/mm3 Luminal A Basal-like | 7.5 (−6.1–21.2) | >0.05 | ||
| pMVD, microvessels/mm3 Luminal A Basal-like | 1.9 (0.7– 3.1) | 0.002 * | ||
| VPI, percentage points Luminal A Basal-like | 1.7 (0.3–3) | 0.014 * | ||
| MVD, microvessels/mm3 Luminal A per 10 vessels increase Basal-like per 10 vessels increase | 1.22 (1.09–1.37) 1.04 (0.95–11.5) | <0.001 * >0.05 | ||
| pMVD, microvessels/mm3 Luminal A per 1 vessel increase Basal-like per 1 vessel increase | 1.11 (0.86–1.43) 1.04 (0.96–1.14) | >0.05 >0.05 | ||
| VPI, percentage points Luminal A per % point increase Basal-like per % point increase | 0.98 (0.83–1.16) 1.02 (0.91–1.13) | >0.05 >0.05 |
| Cytokine | Levels | Environment | Function | Impact on Prognosis |
|---|---|---|---|---|
| TGFβ | ++ | tumor/serum | enhances tumor vascularity, promotes immune evasion and ECM degradation | early relapse and metastases worse survival |
| IL-1β | ++ | tumor/serum | enhances tumor vascularity, inhibits apoptosis in cancer cells, downregulates ER | not determined |
| IL-6 | ++ | serum | promotes EMT and tumor aggressiveness (inhibits response to chemotherapy) | worse survival |
| TNFα | ++ | serum | inhibits apoptosis in cancer cells | not determined |
| IL-8 | ++ | serum | enhances endothelial cell proliferation and MMP production | not determined |
| Leptin | ++ | serum | promotes breast carcinogenesis | not determined |
| IL-10 | ++ | serum | promotes immune evasion | not determined |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Andrea, M.R.; Cereda, V.; Coppola, L.; Giordano, G.; Remo, A.; De Santis, E. Propensity for Early Metastatic Spread in Breast Cancer: Role of Tumor Vascularization Features and Tumor Immune Infiltrate. Cancers 2021, 13, 5917. https://doi.org/10.3390/cancers13235917
D’Andrea MR, Cereda V, Coppola L, Giordano G, Remo A, De Santis E. Propensity for Early Metastatic Spread in Breast Cancer: Role of Tumor Vascularization Features and Tumor Immune Infiltrate. Cancers. 2021; 13(23):5917. https://doi.org/10.3390/cancers13235917
Chicago/Turabian StyleD’Andrea, Mario Rosario, Vittore Cereda, Luigi Coppola, Guido Giordano, Andrea Remo, and Elena De Santis. 2021. "Propensity for Early Metastatic Spread in Breast Cancer: Role of Tumor Vascularization Features and Tumor Immune Infiltrate" Cancers 13, no. 23: 5917. https://doi.org/10.3390/cancers13235917
APA StyleD’Andrea, M. R., Cereda, V., Coppola, L., Giordano, G., Remo, A., & De Santis, E. (2021). Propensity for Early Metastatic Spread in Breast Cancer: Role of Tumor Vascularization Features and Tumor Immune Infiltrate. Cancers, 13(23), 5917. https://doi.org/10.3390/cancers13235917