
Immunotherapeutic Targeting of Mesothelin Positive Pediatric AML Using Bispecific T Cell Engaging Antibodies

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell and Patient-Derived Xenograft Lines
2.2. Generation of an MV4;11 Cell Line Expressing MSLN (MV4;11:MSLN)
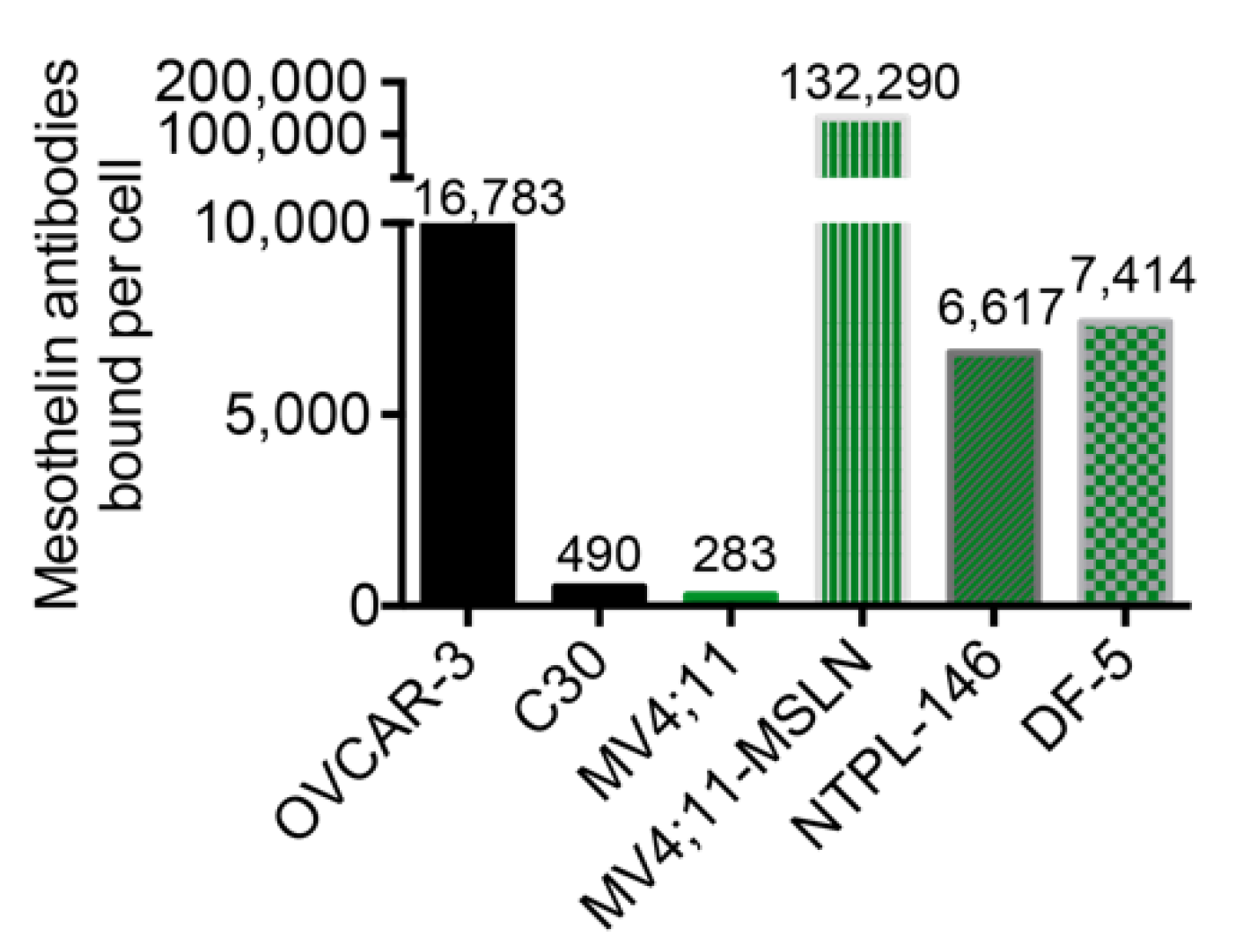
2.3. Flow Cytometry and Quantitation of MSLN Cell Surface Expression
2.4. BsAb Design, Protein Expression and Purification from Human Cells
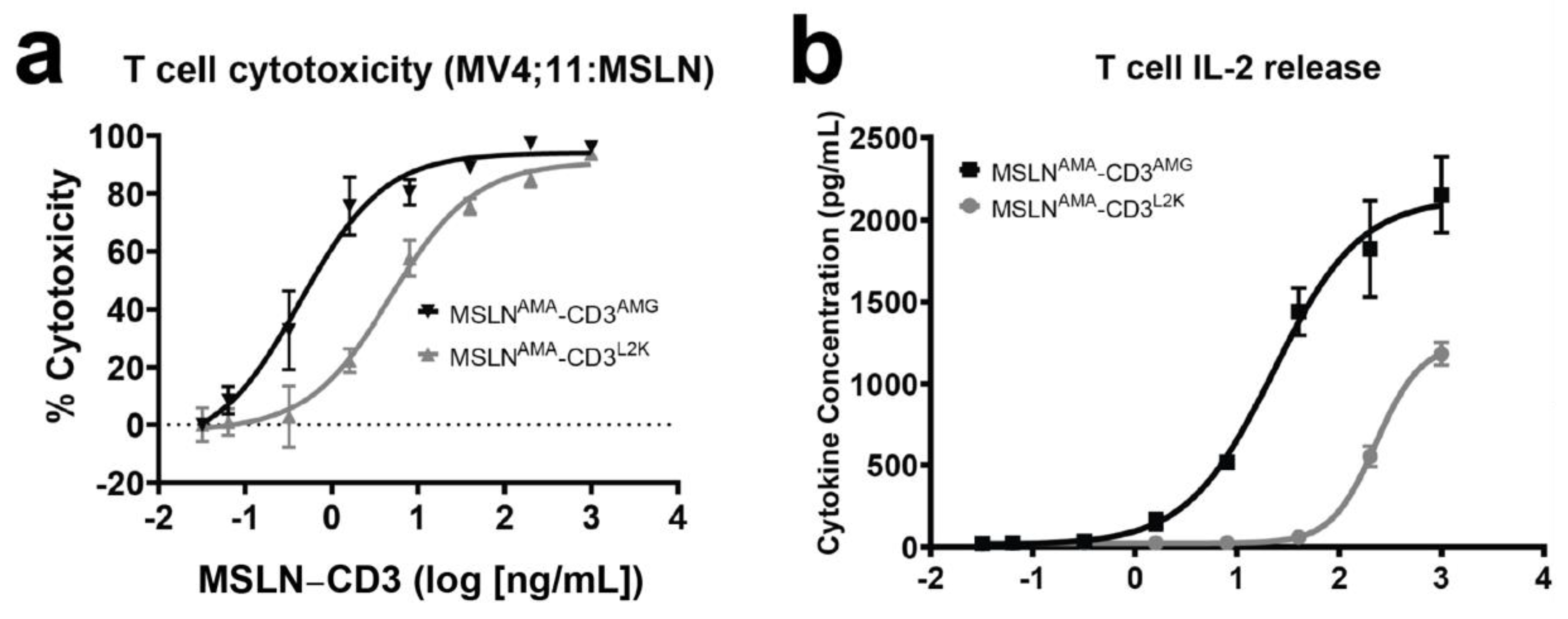
2.5. T Cell Cytotoxicity
2.6. Cytokine Detection and Quantification
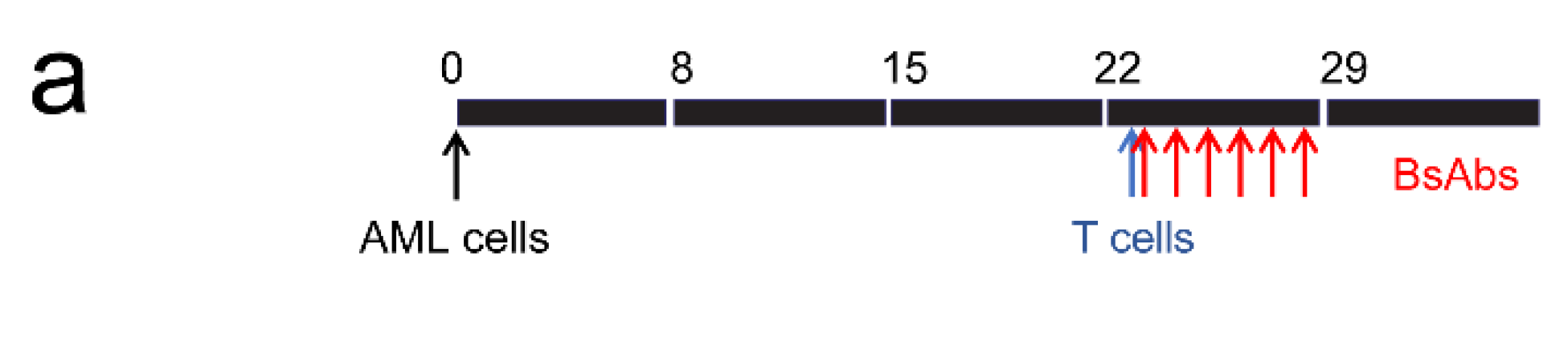
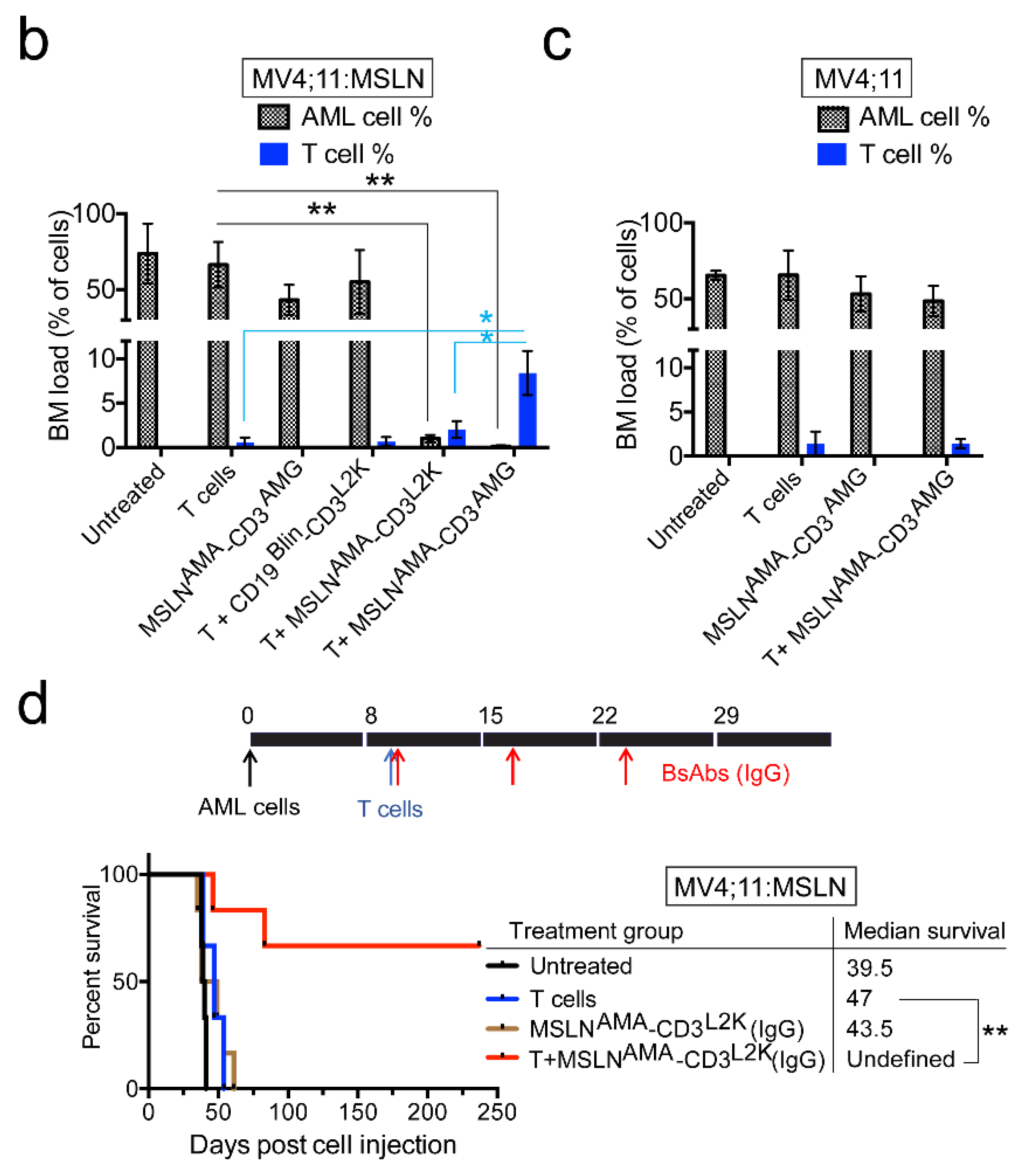
2.7. Leukemia Xenograft Models
2.8. Statistical Analysis
3. Results
3.1. Design and Expression of BsAbs Co-Targeting MSLN and CD3
3.2. Evaluation of In Vitro Activity of MSLN-Targeting BsAbs
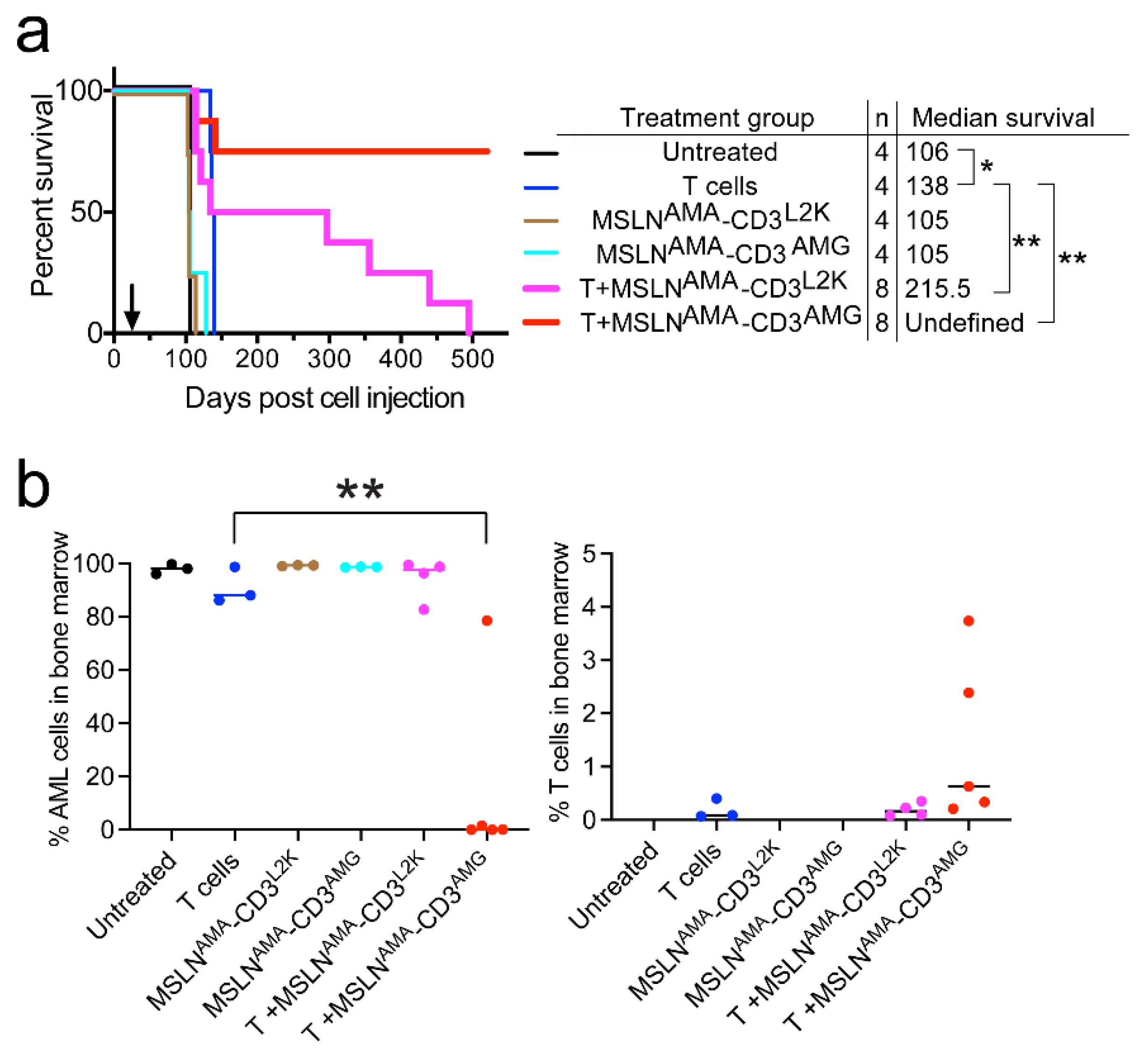
3.3. Testing MSLN-Targeting BsAbs in Cell Line-Derived Xenograft Models with or without MSLN Expression
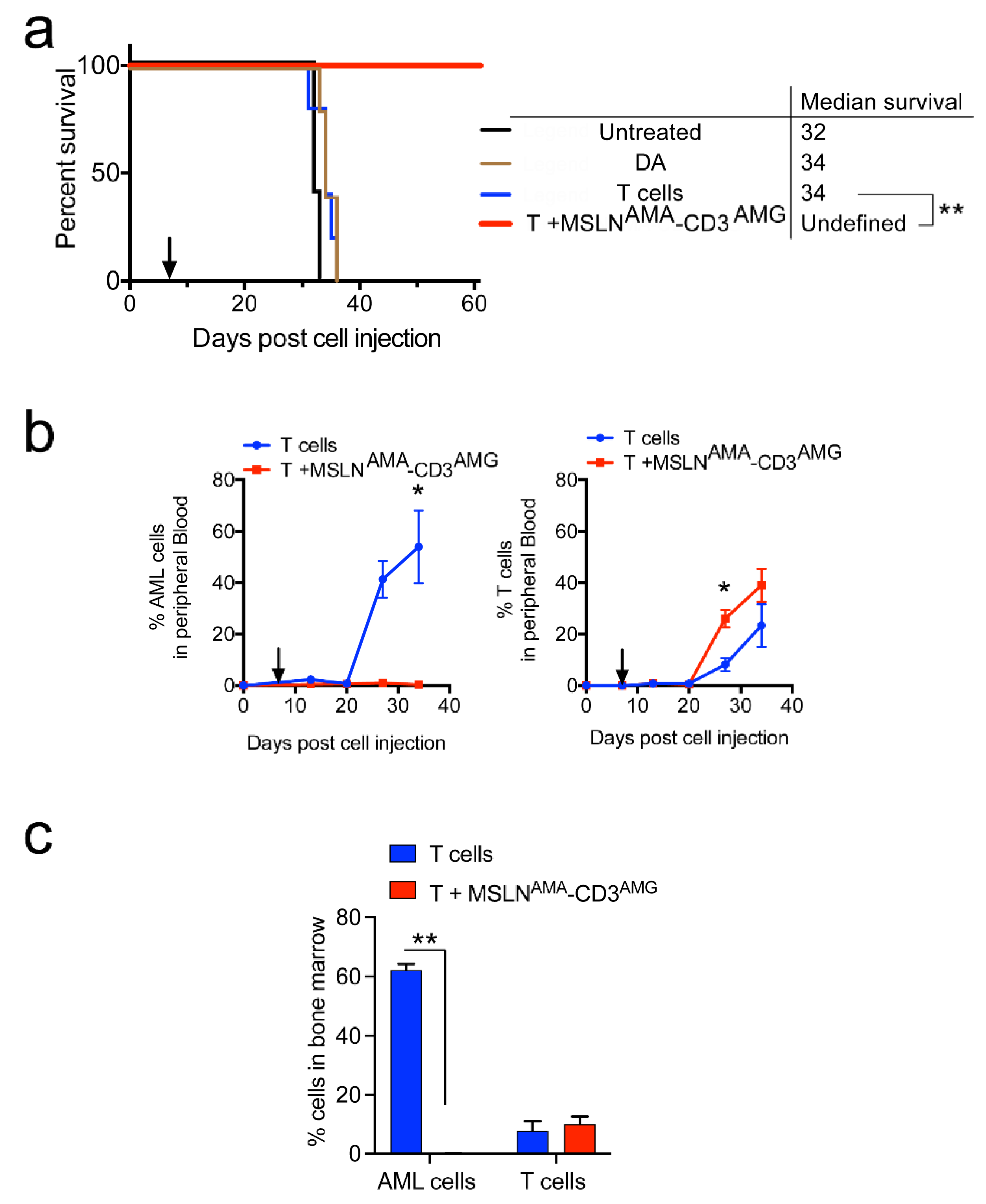
3.4. Testing MSLN-Targeting BsAbs in Patient-Derived Xenograft Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conneely, S.E.; Rau, R.E. The genomics of acute myeloid leukemia in children. Cancer Metastasis Rev. 2020, 39, 189–209. [Google Scholar] [CrossRef]
- Jones, L.; McCarthy, P.; Bond, J. Epigenetics of paediatric acute myeloid leukaemia. Br. J. Haematol. 2020, 188, 63–76. [Google Scholar] [CrossRef]
- Tarlock, K.; Zhong, S.; He, Y.; Ries, R.; Severson, E.; Bailey, M.; Morley, S.; Balasubramanian, S.; Erlich, R.; Lipson, D.; et al. Distinct age-associated molecular profiles in acute myeloid leukemia defined by comprehensive clinical genomic profiling. Oncotarget 2018, 9, 26417–26430. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Pui, C.H. Immunotherapy in pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev. 2019, 38, 595–610. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Velasquez, M.P.; Gottschalk, S. Advances in immunotherapy for pediatric acute myeloid leukemia. Expert Opin. Biol. Ther. 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Lamble, A.J.; Tasian, S.K. Opportunities for immunotherapy in childhood acute myeloid leukemia. Blood Adv. 2019, 3, 3750–3758. [Google Scholar] [CrossRef]
- Steinbach, D.; Schramm, A.; Eggert, A.; Onda, M.; Dawczynski, K.; Rump, A.; Pastan, I.; Wittig, S.; Pfaffendorf, N.; Voigt, A.; et al. Identification of a set of seven genes for the monitoring of minimal residual disease in pediatric acute myeloid leukemia. Clin. Cancer Res. 2006, 12, 2434–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarlock, K.; Kaeding, A.J.; Alonzo, T.A.; Loken, M.R.; Ries, R.E.; Pardo, L.; Gerbing, R.; Farrar, J.E.; Guidry Auvil, J.M.; Gerhard, D.S.; et al. Discovery and Validation of Cell-Surface Protein Mesothelin (MSLN) as a Novel Therapeutic Target in AML: Results from the COG/NCI Target AML Initiative. Blood 2016, 128, 2873. [Google Scholar] [CrossRef]
- Steinbach, D.; Onda, M.; Voigt, A.; Dawczynski, K.; Wittig, S.; Hassan, R.; Gruhn, B.; Pastan, I. Mesothelin, a possible target for immunotherapy, is expressed in primary AML cells. Eur. J. Haematol. 2007, 79, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Cohen, S.J.; Phillips, M.; Pastan, I.; Sharon, E.; Kelly, R.J.; Schweizer, C.; Weil, S.; Laheru, D. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin. Cancer Res. 2010, 16, 6132–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quanz, M.; Hagemann, U.B.; Zitzmann-Kolbe, S.; Stelte-Ludwig, B.; Golfier, S.; Elbi, C.; Mumberg, D.; Ziegelbauer, K.; Schatz, C.A. Anetumab ravtansine inhibits tumor growth and shows additive effect in combination with targeted agents and chemotherapy in mesothelin-expressing human ovarian cancer models. Oncotarget 2018, 9, 34103–34121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, U.B.; Ellingsen, C.; Schuhmacher, J.; Kristian, A.; Mobergslien, A.; Cruciani, V.; Wickstroem, K.; Schatz, C.A.; Kneip, C.; Golfier, S.; et al. Mesothelin-Targeted Thorium-227 Conjugate (MSLN-TTC): Preclinical Evaluation of a New Targeted Alpha Therapy for Mesothelin-Positive Cancers. Clin. Cancer Res. 2019, 25, 4723–4734. [Google Scholar] [CrossRef] [Green Version]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef] [Green Version]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Yoon, A.; Lee, S.; Lee, S.; Lim, S.; Park, Y.Y.; Song, E.; Kim, D.S.; Kim, K.; Lim, Y. A Novel T Cell-Engaging Bispecific Antibody for Treating Mesothelin-Positive Solid Tumors. Biomolecules 2020, 10, 399. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Hu, Y.; Nguyen, C.; Yan, C.; Ries, R.E.; Bolouri, H.; Smith, J.L.; Chen, Q.; Kolb, E.A.; Meshinchi, S.; et al. Altered Transcriptome Uniquely Associated with AML Vs. Normal Hematopoiesis. Blood 2017, 130, 3792. [Google Scholar] [CrossRef]
- Von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnapillai, A.; Kolb, E.A.; McCahan, S.M.; Barwe, S.P. Epigenetic drug combination induces remission in mouse xenograft models of pediatric acute myeloid leukemia. Leuk. Res. 2017, 58, 91–97. [Google Scholar] [CrossRef]
- Gopalakrishnapillai, A.; Kolb, E.A.; Dhanan, P.; Bojja, A.S.; Mason, R.W.; Corao, D.; Barwe, S.P. Generation of Pediatric Leukemia Xenograft Models in NSG-B2m Mice: Comparison with NOD/SCID Mice. Front. Oncol. 2016, 6, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, E.C.; Murakami, M.A.; Christodoulou, A.; Christie, A.L.; Koster, J.; DeSouza, T.A.; Morgan, E.A.; Kallgren, S.P.; Liu, H.; Wu, S.C.; et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell 2016, 29, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Bandaranayake, A.D.; Correnti, C.; Ryu, B.Y.; Brault, M.; Strong, R.K.; Rawlings, D.J. Daedalus: A robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res. 2011, 39, e143. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.S.; Pastan, I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol. 1999, 17, 568–572. [Google Scholar] [CrossRef]
- Hassan, R.; Ebel, W.; Routhier, E.L.; Patel, R.; Kline, J.B.; Zhang, J.; Chao, Q.; Jacob, S.; Turchin, H.; Gibbs, L.; et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007, 7, 20. [Google Scholar]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M.; et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol. Cancer Ther. 2014, 13, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.K.; Kung Sutherland, M.; Li, Y.; Zalevsky, J.; Schell, S.; Leung, W. Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin. Cancer Res. 2012, 18, 6296–6305. [Google Scholar] [CrossRef] [Green Version]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Von Neuhoff, C.; Reinhardt, D.; Sander, A.; Zimmermann, M.; Bradtke, J.; Betts, D.R.; Zemanova, Z.; Stary, J.; Bourquin, J.P.; Haas, O.A.; et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J. Clin. Oncol. 2010, 28, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Agerstam, H.; Karlsson, C.; Hansen, N.; Sanden, C.; Askmyr, M.; von Palffy, S.; Hogberg, C.; Rissler, M.; Wunderlich, M.; Juliusson, G.; et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2015, 112, 10786–10791. [Google Scholar] [CrossRef] [Green Version]
- Stove, H.K.; Sandahl, J.D.; Abrahamsson, J.; Asdahl, P.H.; Forestier, E.; Ha, S.Y.; Jahnukainen, K.; Jonsson, O.G.; Lausen, B.; Palle, J.; et al. Extramedullary leukemia in children with acute myeloid leukemia: A population-based cohort study from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjer-Nielsen, L.; Dunstone, M.A.; Kostenko, L.; Ely, L.K.; Beddoe, T.; Mifsud, N.A.; Purcell, A.W.; Brooks, A.G.; McCluskey, J.; Rossjohn, J. Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. USA 2004, 101, 7675–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Takeuchi, K.; Sun, Z.Y.; Touma, M.; Castro, C.E.; Fahmy, A.; Lang, M.J.; Wagner, G.; Reinherz, E.L. The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 2009, 284, 31028–31037. [Google Scholar] [CrossRef] [Green Version]
- Przespolewski, A.C.; Griffiths, E.A. BITES and CARS and checkpoints, oh my! Updates regarding immunotherapy for myeloid malignancies from the 2018 annual ASH meeting. Blood Rev. 2020, 43, 100654. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopalakrishnapillai, A.; Correnti, C.E.; Pilat, K.; Lin, I.; Chan, M.K.; Bandaranayake, A.D.; Mehlin, C.; Kisielewski, A.; Hamill, D.; Kaeding, A.J.; et al. Immunotherapeutic Targeting of Mesothelin Positive Pediatric AML Using Bispecific T Cell Engaging Antibodies. Cancers 2021, 13, 5964. https://doi.org/10.3390/cancers13235964
Gopalakrishnapillai A, Correnti CE, Pilat K, Lin I, Chan MK, Bandaranayake AD, Mehlin C, Kisielewski A, Hamill D, Kaeding AJ, et al. Immunotherapeutic Targeting of Mesothelin Positive Pediatric AML Using Bispecific T Cell Engaging Antibodies. Cancers. 2021; 13(23):5964. https://doi.org/10.3390/cancers13235964
Chicago/Turabian StyleGopalakrishnapillai, Anilkumar, Colin E. Correnti, Kristina Pilat, Ida Lin, Man Kid Chan, Ashok D. Bandaranayake, Christopher Mehlin, Anne Kisielewski, Darcy Hamill, Allison J. Kaeding, and et al. 2021. "Immunotherapeutic Targeting of Mesothelin Positive Pediatric AML Using Bispecific T Cell Engaging Antibodies" Cancers 13, no. 23: 5964. https://doi.org/10.3390/cancers13235964
APA StyleGopalakrishnapillai, A., Correnti, C. E., Pilat, K., Lin, I., Chan, M. K., Bandaranayake, A. D., Mehlin, C., Kisielewski, A., Hamill, D., Kaeding, A. J., Meshinchi, S., Olson, J. M., Kolb, E. A., & Barwe, S. P. (2021). Immunotherapeutic Targeting of Mesothelin Positive Pediatric AML Using Bispecific T Cell Engaging Antibodies. Cancers, 13(23), 5964. https://doi.org/10.3390/cancers13235964