
Therapeutic Associations Comprising Anti-PD-1/PD-L1 in Breast Cancer: Clinical Challenges and Perspectives

Abstract
:Simple Summary
Abstract
1. Background and Current Development of Immunotherapy with Anti-PD-1/PD-L1 in Breast Cancer
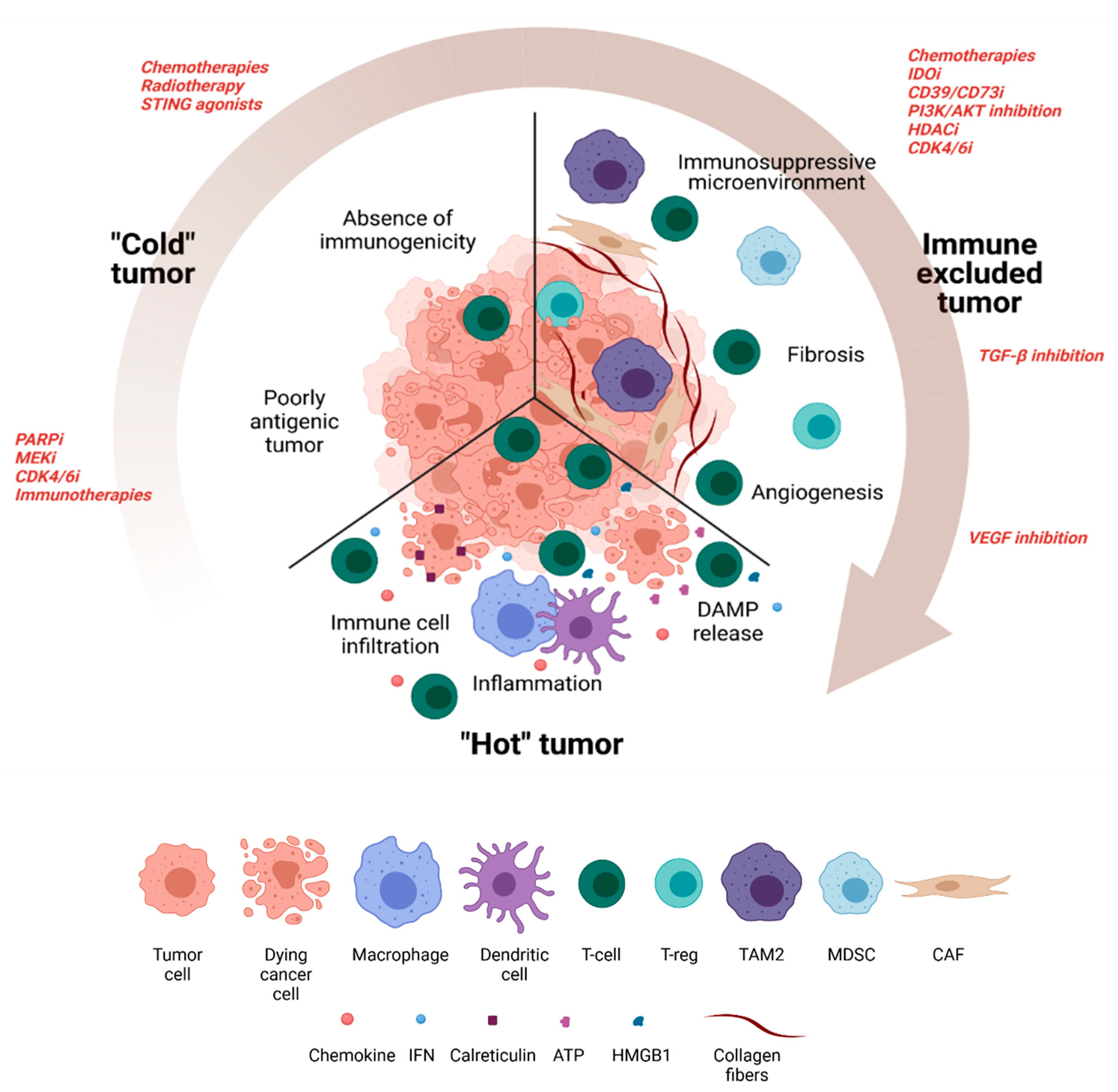
2. Turning a Non-Antigenic and Non-Immunogenic “Cold” BC Tumor into a “Hot” Tumor
2.1. Therapeutic Approaches to Increasing Antigenicity
2.1.1. Poly ADP Ribose Polymerase Inhibitors (PARPi)
2.1.2. MEK Inhibitors
2.1.3. CDK4/6 Inhibitors
2.1.4. Combinations with Other Immunotherapies
2.1.5. Targeting Tumor Antigenicity with Immunological Synergy: The Paradigm of HER-2 Directed Monoclonal Antibodies
2.2. Therapeutic Approaches to Increasing Immunogenicity
2.2.1. Chemotherapies
2.2.2. Radiotherapy
2.2.3. STING Agonists
3. Targeting the Immunosuppressive Microenvironment
3.1. Chemotherapies
3.2. IDO Inhibitors
3.3. The CD39/CD73/Adenosine Pathway
3.4. PI3K/AKT Inhibition
3.5. HDAC Inhibitors
3.6. CDK4/6 Inhibitors
3.7. Autophagy Inhibition
4. Counteract Immune-Excluded Tumors
4.1. Fibrosis
4.2. Tumor Angiogenesis
5. Other Approaches
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast Cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune Checkpoint Inhibitors: Recent Progress and Potential Biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Cruz, L.M.; Czerniecki, B.J. Immunotherapy for Breast Cancer Is Finally at the Doorstep: Immunotherapy in Breast Cancer. Ann. Surg Oncol. 2018, 25, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Gombos, A.; Latifyan, S.; Piccart-Gebhart, M.; Kok, M.; Buisseret, L. Targeting Immune Checkpoints in Breast Cancer: An Update of Early Results. ESMO Open 2017, 2, e000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griguolo, G.; Serna, G.; Pascual, T.; Fasani, R.; Guardia, X.; Chic, N.; Paré, L.; Pernas, S.; Muñoz, M.; Oliveira, M.; et al. Immune Microenvironment Characterisation and Dynamics during Anti-HER2-Based Neoadjuvant Treatment in HER2-Positive Breast Cancer. NPJ Precis. Oncol. 2021, 5, 1–12. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.-L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor Infiltrating Lymphocytes Are Prognostic in Triple Negative Breast Cancer and Predictive for Trastuzumab Benefit in Early Breast Cancer: Results from the FinHER Trial. Ann. Oncol 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical Relevance of Host Immunity in Breast Cancer: From TILs to the Clinic. Nat. Rev Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G Fragment C Receptor Polymorphisms and Clinical Efficacy of Trastuzumab-Based Therapy in Patients with HER-2/Neu-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-Infiltrating Lymphocytes and Prognosis in Different Subtypes of Breast Cancer: A Pooled Analysis of 3771 Patients Treated with Neoadjuvant Therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- He, L.; Wang, Y.; Wu, Q.; Song, Y.; Ma, X.; Zhang, B.; Wang, H.; Huang, Y. Association between Levels of Tumor-Infiltrating Lymphocytes in Different Subtypes of Primary Breast Tumors and Prognostic Outcomes: A Meta-Analysis. BMC Womens Health 2020, 20, 194. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Martin, F.; Ghiringhelli, F. Prognostic Role of FOXP3+ Regulatory T Cells Infiltrating Human Carcinomas: The Paradox of Colorectal Cancer. Cancer Immunol. Immunother. 2011, 60, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Arnould, L.; Apetoh, L.; Coudert, B.; Martin, F.; Chauffert, B.; Fumoleau, P.; Ghiringhelli, F. Pathologic Complete Response to Neoadjuvant Chemotherapy of Breast Carcinoma Is Associated with the Disappearance of Tumor-Infiltrating Foxp3+ Regulatory T Cells. Clin. Cancer Res. 2008, 14, 2413–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Miglietta, F.; Guarneri, V. Immune Infiltrates in Breast Cancer: Recent Updates and Clinical Implications. Cells 2021, 10, 223. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.-P.; et al. Long-Term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor Cell-Intrinsic PD-1 Receptor Is a Tumor Suppressor and Mediates Resistance to PD-1 Blockade Therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cruz, C.; Braiteh, F.S.; Eder, J.P.; Tolaney, S.; Kuter, I.; Nanda, R.; Chung, C.; Cassier, P.; Delord, J.-P.; et al. Abstract 2986: Atezolizumab in Metastatic TNBC (MTNBC): Long-Term Clinical Outcomes and Biomarker Analyses. Cancer Res. 2017, 77, 2986. [Google Scholar]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab Monotherapy for Previously Treated Metastatic Triple-Negative Breast Cancer: Cohort A of the Phase II KEYNOTE-086 Study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, J.; Lipatov, O.; Im, S.-A.; Gonçalves, A.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; Ohtani, S.; et al. LBA21-KEYNOTE-119: Phase III Study of Pembrolizumab (Pembro) versus Single-Agent Chemotherapy (Chemo) for Metastatic Triple Negative Breast Cancer (MTNBC). Ann. Oncol. 2019, 30, v859–v860. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.-T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an Anti-PD-L1 Antibody, in Patients with Locally Advanced or Metastatic Breast Cancer: A Phase 1b JAVELIN Solid Tumor Study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Jammed, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-Induced Immunogenic Modulation of Tumor Enhances Antigen Processing and Calreticulin Exposure, Resulting in Enhanced T-Cell Killing. Oncotarget 2013, 5, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aymeric, L.; Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Martins, I.; Kroemer, G.; Smyth, M.J.; Zitvogel, L. Tumor Cell Death and ATP Release Prime Dendritic Cells and Efficient Anticancer Immunity. Cancer Res. 2010, 70, 855–858. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-Associated Molecular Patterns in Cancer: A Double-Edged Sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Diamond, J.; Hamilton, E.; Pohlmann, P.; Tolaney, S.; Chang, C.-W.; Zhang, W.; Iizuka, K.; Foster, P.; Molinero, L.; et al. Atezolizumab Plus Nab-Paclitaxel in the Treatment of Metastatic Triple-Negative Breast Cancer With 2-Year Survival Follow-up: A Phase 1b Clinical Trial. JAMA Oncol. 2018, 5, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolaney, S.M.; Barroso-Sousa, R.; Keenan, T.; Li, T.; Trippa, L.; Vaz-Luis, I.; Wulf, G.; Spring, L.; Sinclair, N.F.; Andrews, C.; et al. Effect of Eribulin with or without Pembrolizumab on Progression-Free Survival for Patients With Hormone Receptor-Positive, ERBB2-Negative Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1598–1605. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary Results from IMpassion131, a Double-Blind, Placebo-Controlled, Randomised Phase III Trial of First-Line Paclitaxel with or without Atezolizumab for Unresectable Locally Advanced/Metastatic Triple-Negative Breast Cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus Chemotherapy versus Placebo plus Chemotherapy for Previously Untreated Locally Recurrent Inoperable or Metastatic Triple-Negative Breast Cancer (KEYNOTE-355): A Randomised, Placebo-Controlled, Double-Blind, Phase 3 Clinical Trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Schmid, P.; Salgado, R.; Park, Y.H.; Muñoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.-A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus Chemotherapy as Neoadjuvant Treatment of High-Risk, Early-Stage Triple-Negative Breast Cancer: Results from the Phase 1b Open-Label, Multicohort KEYNOTE-173 Study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.-U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A Randomised Phase II Study Investigating Durvalumab in Addition to an Anthracycline Taxane-Based Neoadjuvant Therapy in Early Triple-Negative Breast Cancer: Clinical Results and Biomarker Analysis of GeparNuevo Study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Schneeweiss, A.; Huober, J.B.; Braun, M.; Rey, J.; Blohmer, J.U.; Furlanetto, J.; Zahm, D.M.; Hanusch, C.; Thomalla, J.; et al. Durvalumab Improves Long-Term Outcome in TNBC: Results from the Phase II Randomized GeparNUEVO Study Investigating Neodjuvant Durvalumab in Addition to an Anthracycline/Taxane Based Neoadjuvant Chemotherapy in Early Triple-Negative Breast Cancer (TNBC). JCO 2021, 39, 506. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant Atezolizumab in Combination with Sequential Nab-Paclitaxel and Anthracycline-Based Chemotherapy versus Placebo and Chemotherapy in Patients with Early-Stage Triple-Negative Breast Cancer (IMpassion031): A Randomised, Double-Blind, Phase 3 Trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Gianni, L.; Huang, C.-S.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Bianchini, G.; Russo, S.; et al. Abstract GS3-04: Pathologic Complete Response (PCR) to Neoadjuvant Treatment with or without Atezolizumab in Triple Negative, Early High-Risk and Locally Advanced Breast Cancer. NeoTRIPaPDL1 Michelangelo Randomized Study. Cancer Res. 2020, 80, GS3-04. [Google Scholar]
- Lüönd, F.; Tiede, S.; Christofori, G. Breast Cancer as an Example of Tumour Heterogeneity and Tumour Cell Plasticity during Malignant Progression. Br. J. Cancer 2021, 1–12. [Google Scholar] [CrossRef]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 289. [Google Scholar] [CrossRef]
- Karn, T.; Jiang, T.; Hatzis, C.; Sänger, N.; El-Balat, A.; Rody, A.; Holtrich, U.; Becker, S.; Bianchini, G.; Pusztai, L. Association Between Genomic Metrics and Immune Infiltration in Triple-Negative Breast Cancer. JAMA Oncol. 2017, 3, 1707–1711. [Google Scholar] [CrossRef]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.B.; Patwardhan, G.A.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological Differences between Primary and Metastatic Breast Cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.-Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-Induced Immunogenic Modulation of Tumor Cells Enhances Killing by Cytotoxic T Lymphocytes and Is Distinct from Immunogenic Cell Death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune Induction Strategies in Metastatic Triple-Negative Breast Cancer to Enhance the Sensitivity to PD-1 Blockade: The TONIC Trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Esteva, F.J.; Hubbard-Lucey, V.M.; Tang, J.; Pusztai, L. Immunotherapy and Targeted Therapy Combinations in Metastatic Breast Cancer. Lancet Oncol. 2019, 20, e175–e186. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and Mutational Determinants of High Tumor Mutation Burden in Breast Cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Kronbichler, A.; Eisenhut, M.; Hong, S.H.; van der Vliet, H.J.; Kang, J.; Shin, J.I.; Gamerith, G. Tumor Mutational Burden and Efficacy of Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De kruijf, E.; van Nes, J.; Sajet, A.; Tummers, Q.; Putter, H.; Osanto, S.; Speetjens, F.; Smit, V.; Liefers, G.-J.; Velde, C.; et al. The Predictive Value of HLA Class I Tumor Cell Expression and Presence of Intratumoral Tregs for Chemotherapy in Patients with Early Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1272–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-Tumor Genomic Biomarkers for PD-1 Checkpoint Blockade-Based Immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.X.; Leong, C.-O. Association of BRCA1- and BRCA2-Deficiency with Mutation Burden, Expression of PD-L1/PD-1, Immune Infiltrates, and T Cell-Inflamed Signature in Breast Cancer. PLoS ONE 2019, 14, e0215381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and Durvalumab in Patients with Germline BRCA-Mutated Metastatic Breast Cancer (MEDIOLA): An Open-Label, Multicentre, Phase 1/2, Basket Study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.S.; Mita, M.M.; McCann, G.A.-L.; Tan, A.R.; Wahner Hendrickson, A.E.; Forero-Torres, A.; Anders, C.K.; Wulf, G.M.; et al. TOPACIO/Keynote-162: Niraparib + Pembrolizumab in Patients (Pts) with Metastatic Triple-Negative Breast Cancer (TNBC), a Phase 2 Trial. JCO 2018, 36, 1011. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Faja, L.R.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.; Comin-Anduix, B.; et al. Improved Antitumor Activity of Immunotherapy with BRAF and MEK Inhibitors in BRAFV600E Melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-Tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, D.A.; James, J.L.; Axelrod, M.L.; Balko, J.M. MEK Inhibition Activates STAT Signaling to Increase Breast Cancer Immunogenicity via MHC-I Expression. Cancer Drug Resist. 2020, 3, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Brufsky, A.; Kim, S.B.; Zvirbule, Ž.; Eniu, A.; Mebis, J.; Sohn, J.H.; Wongchenko, M.; Chohan, S.; Amin, R.; Yan, Y.; et al. A Phase II Randomized Trial of Cobimetinib plus Chemotherapy, with or without Atezolizumab, as First-Line Treatment for Patients with Locally Advanced or Metastatic Triple-Negative Breast Cancer (COLET): Primary Analysis. Ann. Oncol. 2021, 32, 652–660. [Google Scholar] [CrossRef]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist Immunotherapy Restores T Cell Function Following MEK Inhibition Improving Efficacy in Breast Cancer. Nat. Commun. 2017, 8, 606. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Forster, M.D.; Summers, Y.J.; Good, J.; Sarker, S.-J.; Lim, L.; Mousa, K.; Middleton, G.W. A Study of Vistusertib in Combination with Selumetinib in Patients with Advanced Cancers: TORCMEK Phase Ib Results. JCO 2017, 35, 2548. [Google Scholar] [CrossRef]
- Li, J.; Huo, X.; Zhao, F.; Ren, D.; Ahmad, R.; Yuan, X.; Du, F.; Zhao, J. Association of Cyclin-Dependent Kinases 4 and 6 Inhibitors With Survival in Patients With Hormone Receptor-Positive Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2020, 3, e2020312. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-Cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.Y.; Sorrentino, J.A.; Dragnev, K.H.; Weiss, J.M.; Owonikoko, T.K.; Rytlewski, J.A.; Hood, J.; Yang, Z.; Malik, R.K.; Strum, J.C.; et al. CDK4/6 Inhibition Enhances Antitumor Efficacy of Chemotherapy and Immune Checkpoint Inhibitor Combinations in Preclinical Models and Enhances T-Cell Activation in Patients with SCLC Receiving Chemotherapy. J. Immunother. Cancer 2020, 8, e000847. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Kabos, P.; Beck, J.T.; Chisamore, M.J.; Hossain, A.; Chen, Y.; Tolaney, S.M. A Phase Ib Study of Abemaciclib in Combination with Pembrolizumab for Patients with Hormone Receptor Positive (HR+), Human Epidermal Growth Factor Receptor 2 Negative (HER2-) Locally Advanced or Metastatic Breast Cancer (MBC) (NCT02779751): Interim Results. JCO 2020, 38, 1051. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Larkin, J.; Tabernero, J.; Bonini, C. Enhancing Anti-Tumour Efficacy with Immunotherapy Combinations. Int. J. STD AIDS 2021, 397, 1010–1022. [Google Scholar] [CrossRef]
- Chikuma, S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. Curr Top. Microbiol. Immunol. 2017, 410, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, C.A.; Kato, T.; Park, J.-H.; Kiyotani, K.; Rademaker, A.; Shah, A.N.; Gross, L.; Blanco, L.Z.; Jain, S.; Flaum, L.; et al. A Pilot Study of Durvalumab and Tremelimumab and Immunogenomic Dynamics in Metastatic Breast Cancer. Oncotarget 2018, 9, 18985–18996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlén, E.; Veitonmäki, N.; Norlén, P. Bispecific Antibodies in Cancer Immunotherapy. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Gennari, R.; Menard, S.; Fagnoni, F.; Ponchio, L.; Scelsi, M.; Tagliabue, E.; Castiglioni, F.; Villani, L.; Magalotti, C.; Gibelli, N.; et al. Pilot Study of the Mechanism of Action of Preoperative Trastuzumab in Patients with Primary Operable Breast Tumors Overexpressing HER2. Clin. Cancer Res. 2004, 10, 5650–5655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-Based Treatment of HER2-Positive Breast Cancer: An Antibody-Dependent Cellular Cytotoxicity Mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Holgado, E.; Perez-Garcia, J.; Gion, M.; Cortes, J. Is There a Role for Immunotherapy in HER2-Positive Breast Cancer? NPJ Breast Cancer 2018, 4, 21. [Google Scholar] [CrossRef]
- Müller, P.; Kreuzaler, M.; Khan, T.; Thommen, D.S.; Martin, K.; Glatz, K.; Savic, S.; Harbeck, N.; Nitz, U.; Gluz, O.; et al. Trastuzumab Emtansine (T-DM1) Renders HER2+ Breast Cancer Highly Susceptible to CTLA-4/PD-1 Blockade. Sci. Transl. Med. 2015, 7, 315ra188. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti–ErbB-2 MAb Therapy Requires Type I and II Interferons and Synergizes with Anti–PD-1 or Anti-CD137 MAb Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus Trastuzumab in Trastuzumab-Resistant, Advanced, HER2-Positive Breast Cancer (PANACEA): A Single-Arm, Multicentre, Phase 1b–2 Trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.-B.; Im, S.-A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab Emtansine plus Atezolizumab versus Trastuzumab Emtansine plus Placebo in Previously Treated, HER2-Positive Advanced Breast Cancer (KATE2): A Phase 2, Multicentre, Randomised, Double-Blind Trial. Lancet Oncol. 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Mignot, G.; Panaretakis, T.; Kroemer, G.; Zitvogel, L. Immunogenicity of Anthracyclines: Moving towards More Personalized Medicine. Trends Mol. Med. 2008, 14, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Mimura, K.; Okayama, H.; Saito, M.; Chida, S.; Noda, M.; Nakajima, T.; Saito, K.; Abe, N.; Ohki, S.; et al. Immunogenic Tumor Cell Death Induced by Chemotherapy in Patients with Breast Cancer and Esophageal Squamous Cell Carcinoma. Oncol. Rep. 2018, 39, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Enot, D.; Senovilla, L.; Ghiringhelli, F.; Poirier-Colame, V.; Chaba, K.; Semeraro, M.; Chaix, M.; Penault-Llorca, F.; Arnould, L.; et al. The Presence of LC3B Puncta and HMGB1 Expression in Malignant Cells Correlate with the Immune Infiltrate in Breast Cancer. Autophagy 2016, 12, 864–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, L.; Menzel, U.; Prummer, M.; Müller, P.; Buchi, M.; Kashyap, A.; Haessler, U.; Yermanos, A.; Gébleux, R.; Briendl, M.; et al. A Novel Anti-HER2 Anthracycline-Based Antibody-Drug Conjugate Induces Adaptive Anti-Tumor Immunity and Potentiates PD-1 Blockade in Breast Cancer. J. Immunother. Cancer 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Gebremeskel, S.; Lobert, L.; Tanner, K.; Walker, B.; Oliphant, T.; Clarke, L.E.; Dellaire, G.; Johnston, B. Natural Killer T-Cell Immunotherapy in Combination with Chemotherapy-Induced Immunogenic Cell Death Targets Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-Analysis of Tumor- and T Cell-Intrinsic Mechanisms of Sensitization to Checkpoint Inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef] [PubMed]
- Ludgate, C.M. Optimizing Cancer Treatments to Induce an Acute Immune Response: Radiation Abscopal Effects, PAMPs, and DAMPs. Clin. Cancer Res. 2012, 18, 4522–4525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and Dual Checkpoint Blockade Activate Non-Redundant Immune Mechanisms in Cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Abbassi, L.; Romano, E.; Kirova, Y. Radiation Therapy and Immunotherapy in Breast Cancer Treatment: Preliminary Data and Perspectives. Expert Rev. Anticancer 2021, 21, 501–510. [Google Scholar] [CrossRef]
- Ho, A.Y.; Barker, C.A.; Arnold, B.B.; Powell, S.N.; Hu, Z.I.; Gucalp, A.; Lebron-Zapata, L.; Wen, H.Y.; Kallman, C.; D’Agnolo, A.; et al. A Phase 2 Clinical Trial assessing the efficacy and Safety of Pembrolizumab and Radiotherapy in Patients with Metastatic Triple-Negative Breast Cancer. Cancer 2020, 126, 850–860. [Google Scholar] [CrossRef]
- Hu, Z.I.; McArthur, H.L.; Ho, A.Y. The Abscopal Effect of Radiation Therapy: What Is It and How Can We Use It in Breast Cancer? Curr. Breast Cancer Rep. 2017, 9, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocenzi, T.; Cottam, B.; Newell, P.; Wolf, R.F.; Hansen, P.D.; Hammill, C.; Solhjem, M.C.; To, Y.-Y.; Greathouse, A.; Tormoen, G.; et al. A Hypofractionated Radiation Regimen Avoids the Lymphopenia Associated with Neoadjuvant Chemoradiation Therapy of Borderline Resectable and Locally Advanced Pancreatic Adenocarcinoma. J. Immunother. Cancer 2016, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Morisada, M.; Clavijo, P.E.; Moore, E.; Sun, L.; Chamberlin, M.; Van Waes, C.; Hodge, J.W.; Mitchell, J.B.; Friedman, J.; Allen, C.T. PD-1 Blockade Reverses Adaptive Immune Resistance Induced by High-Dose Hypofractionated but Not Low-Dose Daily Fractionated Radiation. Oncoimmunology 2018, 7, e1395996. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Coleman, C.N.; Formenti, S.C. Radiotherapy: Changing the Game in Immunotherapy. Trends Cancer 2016, 2, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrales, L.; Gajewski, T.F. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin. Cancer Res. 2015, 21, 4774–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Watkins-Schulz, R.; Junkins, R.D.; David, C.N.; Johnson, B.M.; Montgomery, S.A.; Peine, K.J.; Darr, D.B.; Yuan, H.; McKinnon, K.P.; et al. A Nanoparticle-Incorporated STING Activator Enhances Antitumor Immunity in PD-L1-Insensitive Models of Triple-Negative Breast Cancer. JCI Insight 2018, 3, 120638. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. CGAS Is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Jadhav, H.; Kothari, A.; Liu, R.; Guerriero, J.L.; Shapiro, G.I. STING Agonism Enhances Anti-Tumor Immune Responses and Therapeutic Efficacy of PARP Inhibition in BRCA-Associated Breast Cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ Regulatory T Cells Suppress Tumor Immunity but Are Sensitive to Cyclophosphamide Which Allows Immunotherapy of Established Tumors to Be Curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil Selectively Kills Tumor-Associated Myeloid-Derived Suppressor Cells Resulting in Enhanced T Cell-Dependent Antitumor Immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.A.; Ponce de Léon, J.L.; Benguigui, M.; Xu, P.; Chow, A.; Cruz-Muñoz, W.; Man, S.; Shaked, Y.; Kerbel, R.S. Immunostimulatory and Anti-Tumor Metronomic Cyclophosphamide Regimens Assessed in Primary Orthotopic and Metastatic Murine Breast Cancer. NPJ Breast Cancer 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, T.; Shurin, G.V.; Naiditch, H.; Sevko, A.; Umansky, V.; Shurin, M.R. Paclitaxel Promotes Differentiation of Myeloid-Derived Suppressor Cells into Dendritic Cells in Vitro in a TLR4-Independent Manner. J. Immunotoxicol. 2012, 9, 292–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H. Indoleamine 2,3-Dioxygenase, Tregs and Cancer. Curr. Med. Chem. 2011, 18, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Jackson, E.; Neuger, T.; Dees, E.C.; Harvey, R.D.; Han, H.; Ismail-Khan, R.; Minton, S.; Vahanian, N.N.; Link, C.; et al. A First in Man Phase I Trial of the Oral Immunomodulator, Indoximod, Combined with Docetaxel in Patients with Metastatic Solid Tumors. Oncotarget 2014, 5, 8136–8146. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). JCO 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A Potent Suppressor of Antitumor Immune Responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A Receptors Potently Suppresses the Metastasis of CD73+ Tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 Promotes Anthracycline Resistance and Poor Prognosis in Triple Negative Breast Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Young, A.; Stannard, K.; Yong, M.; Teng, M.W.L.; Allard, B.; Stagg, J.; Smyth, M.J. Antimetastatic Effects of Blocking PD-1 and the Adenosine A2A Receptor. Cancer Res. 2014, 74, 3652–3658. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-MTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective Inhibition of Regulatory T Cells by Targeting the PI3K–Akt Pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ Is a Molecular Switch That Controls Immune Suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via DsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of Metastatic Mouse Cancers Resistant to Immune Checkpoint Blockade by Suppression of Myeloid-Derived Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Malamas, A.S.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Inhibitors of Histone Deacetylase 1 Reverse the Immune Evasion Phenotype to Enhance T-Cell Mediated Lysis of Prostate and Breast Carcinoma Cells. Oncotarget 2016, 7, 7390–7402. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Lee, M.-J.; Lee, S.; Tomita, S.; Chumsri, S.; Cruickshank, S.; Ordentlich, P.; Trepel, J.B. The Interplay of Epigenetic Therapy and Immunity in Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer: Correlative Analysis of ENCORE 301, a Randomized, Placebo-Controlled Phase II Trial of Exemestane with or without Entinostat. Oncoimmunology 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forero, A.; Stroyakovskiy, D.; Cha, E.; Cruickshank, S.; Hasapidis, J.; Meyers, M.L.; Slamon, D.J. Abstract OT2-01-12: ENCORE 602: A Randomized, Placebo-Controlled, Double-Blind, Multicenter Phase 2 Study (with a Phase 1b Lead-in) of Atezolizumab with or without Entinostat in Patients with Advanced Triple Negative Breast Cancer (ATNBC). Cancer Res. 2017, 77, OT2-01-12. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurvitz, S.; Martin, M.; Abad, M.F.; Chan, D.; Rostorfer, R.; Petru, E.; Barriga, S.; Costigan, T.M.; Caldwell, C.W.; Nguyen, T.; et al. Abstract S4-06: Biological Effects of Abemaciclib in a Phase 2 Neoadjuvant Study for Postmenopausal Patients with HR+, HER2- Breast Cancer. Cancer Res. 2017, 77, S4-S4-06. [Google Scholar] [CrossRef]
- Page, D.B.; Bear, H.; Prabhakaran, S.; Gatti-Mays, M.E.; Thomas, A.; Cobain, E.; McArthur, H.; Balko, J.M.; Gameiro, S.R.; Nanda, R.; et al. Two May Be Better than One: PD-1/PD-L1 Blockade Combination Approaches in Metastatic Breast Cancer. NPJ Breast Cancer 2019, 5, 34. [Google Scholar] [CrossRef]
- Das, M.; Law, S. Role of Tumor Microenvironment in Cancer Stem Cell Chemoresistance and Recurrence. Int. J. Biochem. Cell Biol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Chen, P.; Hsu, W.-H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Farzaneh, M. Signaling Pathways Governing Breast Cancer Stem Cells Behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef]
- Gong, C.; Song, E.; Codogno, P.; Mehrpour, M. The Roles of BECN1 and Autophagy in Cancer Are Context Dependent. Autophagy 2012, 8, 1853–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.; Ahmad, R.; Sharma, S.; Gowrikumar, S.; Primeaux, M.; Rana, S.; Natarajan, A.; Oupicky, D.; Hopkins, C.R.; Dhawan, P.; et al. PIK3C3 Inhibition Promotes Sensitivity to Colon Cancer Therapy by Inhibiting Cancer Stem Cells. Cancers 2021, 13, 2168. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Leone, A.; Piezzo, M.; Caputo, R.; Di Lauro, V.; Di Rella, F.; Fusco, G.; Capozzi, M.; di Gioia, G.; Budillon, A.; et al. Targeting Autophagy in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7836. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Hasmim, M.; Parpal, S.; De Milito, A.; Berchem, G.; Noman, M.Z. Lighting up the Fire in Cold Tumors to Improve Cancer Immunotherapy by Blocking the Activity of the Autophagy-Related Protein PIK3C3/VPS34. Autophagy 2020, 16, 2110–2111. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.; Towers, C.G. Enhancing Anti-Tumor Immunity by Autophagy Inhibition. Nat. Cancer 2021, 2, 484–486. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothé, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-Associated Fibrosis as a Regulator of Tumor Immunity and Response to Immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and Immune Cells: An Important Regulatory Axis in the Tumor Microenvironment and Progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ Pathway with Galunisertib, a TGFβRI Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination with Checkpoint Blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β Inhibition Enhances Chemotherapy Action against Triple-Negative Breast Cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation And Systemic Transforming Growth Factor β Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced Preclinical Antitumor Activity of M7824, a Bifunctional Fusion Protein Simultaneously Targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Madu, C.O.; Wang, S.; Madu, C.O.; Lu, Y. Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment. J. Cancer 2020, 11, 4474–4494. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of Anti-Angiogenic Therapy and Immune Checkpoint Blockade Normalizes Vascular-Immune Crosstalk to Potentiate Cancer Immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar, R.B.; de Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-Vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.-J.; Ooi, C.-H.; Laoui, D.; et al. Dual Angiopoietin-2 and VEGFA Inhibition Elicits Antitumor Immunity That Is Enhanced by PD-1 Checkpoint Blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic Effect of Immune Checkpoint Blockade and Anti-Angiogenesis in Cancer Treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer 2020, 19, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Han, H.H.; Diab, S.; Alemany, C.; Basho, R.; Brown-Glaberman, U.; Meisel, J.; Pluard, T.; Cortes, J.; Dillon, P.; Ettl, J.; et al. Abstract PD1-06: Open Label Phase 1b/2 Study of Ladiratuzumab Vedotin in Combination with Pembrolizumab for First-Line Treatment of Patients with Unresectable Locally-Advanced or Metastatic Triple-Negative Breast Cancer. Cancer Res. 2020, 80, PD1-06. [Google Scholar]



{kind=link}
{kind=link}
| Trial Name/Number | Phase | Subtype/Condition | Immunotherapy Combinations |
|---|---|---|---|
| NCT03219268 | I | HER2/TNBC Advanced Solid Tumors | MGD013:Bispecific antibodies anti-LAG3/anti-PD-1 |
| NCT03440437 | I/II | Advanced Cancer Metastatic Cancer | FS118: Bispecific antibodies anti-LAG3/anti-PD-L1 |
| DUET-2 NCT03517488 | I | Advanced Solid Tumors | XmAb®20717: Bispecific antibodies anti-CTLA4/anti-PD-1 |
| NCT01968109 | I/2a | Advanced Solid Tumors | Nivolumab (anti-PD-1 antibody) + BMS-936558 (anti-LAG3 antibody) |
| AIPAC-002 NCT04252768 | I | Metastatic Breast Cancer HR+ | Eftilagimod Alpha (Soluble LAG-3 Protein) + paclitaxel |
| InCITe NCT03971409 | II | Stage IV or Unresectable TNBC | PF-04518600 (Anti-OX40 antibody) + Avelumab (anti-PD-L1 antibody) |
| 4-1BB/CD137 agonist+ avelumab | |||
| Sacituzumab govitecan (anti-Trop2 antibody) + avelumab | |||
| NCT02794571 | Ia/Ib | Advanced/Metastatic Tumors | Tiragolumab (anti-TIGIT antibody) + Atezolizumab (anti-PD-L1 antibody) + chemotherapies |
| NCT04584112 | Ib | TNBC | Tiragolumab + Atezolizumab + chemotherapies |
| NCT03742349 | I | TNBC | Spartalizumab (anti-PD-1 antibody) + LAG525(anti-LAG-3 antibody) + NIR178 or capmatinib or MCS110 or canakinumab (anti IL-1β antibody) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledys, F.; Kalfeist, L.; Galland, L.; Limagne, E.; Ladoire, S. Therapeutic Associations Comprising Anti-PD-1/PD-L1 in Breast Cancer: Clinical Challenges and Perspectives. Cancers 2021, 13, 5999. https://doi.org/10.3390/cancers13235999
Ledys F, Kalfeist L, Galland L, Limagne E, Ladoire S. Therapeutic Associations Comprising Anti-PD-1/PD-L1 in Breast Cancer: Clinical Challenges and Perspectives. Cancers. 2021; 13(23):5999. https://doi.org/10.3390/cancers13235999
Chicago/Turabian StyleLedys, Fanny, Laura Kalfeist, Loick Galland, Emeric Limagne, and Sylvain Ladoire. 2021. "Therapeutic Associations Comprising Anti-PD-1/PD-L1 in Breast Cancer: Clinical Challenges and Perspectives" Cancers 13, no. 23: 5999. https://doi.org/10.3390/cancers13235999
APA StyleLedys, F., Kalfeist, L., Galland, L., Limagne, E., & Ladoire, S. (2021). Therapeutic Associations Comprising Anti-PD-1/PD-L1 in Breast Cancer: Clinical Challenges and Perspectives. Cancers, 13(23), 5999. https://doi.org/10.3390/cancers13235999