
Impact of DNA Damage Response—Targeted Therapies on the Immune Response to Tumours


Abstract
:Simple Summary
Abstract
1. Introduction
2. Effects of DDR-Targeted Therapies on the Cell Components of the Immune System
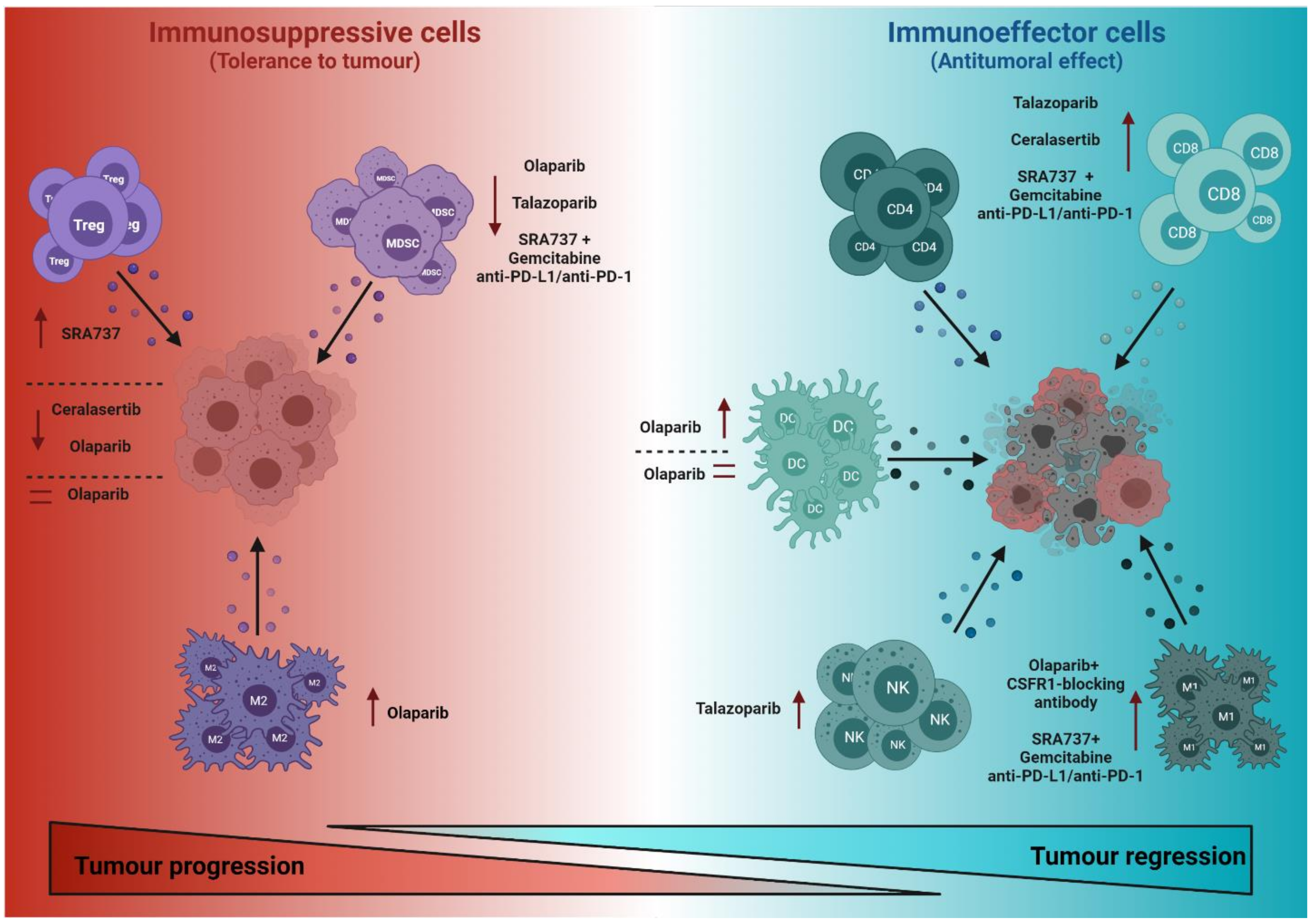
2.1. Effects of DDR-Targeted Therapies in Cells with Immunosuppressive Functions
2.2. Effects of DDR-Targeted Therapies on Immune Cells with Antitumoural Effector Functions
3. DDR-Targeted Agents Modulate Pathways in Tumour Cells That Impact Their Relationships with Immune Cells
3.1. DDR-Targeted Agents Modulate Immune Responses via Activation of the cGAS/STING Pathway in Tumour Cells
3.2. Regulation of the Expression of Immune Checkpoint Interacting Molecules on Tumour Cells by DDR-Targeted Agents
3.3. Modulation of Other Ligands on Tumour Cells by DDR-Targeted Agents That Affect Their Interactions with Immune Cells
4. Concluding Remarks and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tang, M.; Chen, Z.; Nie, L.; Li, S.; Xiong, Y.; Szymonowicz, K.A.; Park, J.-M.; Zhang, H.; Feng, X.; et al. Genetic vulnerabilities upon inhibition of DNA damage response. Nucleic Acids Res. 2021, 49, 8214–8231. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021, 35, 602–618. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685. [Google Scholar] [CrossRef]
- Axelrod, M.L.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Bergman, R.E.; Pilkinton, M.A.; McDonnell, W.J.; Sanchez, V.; Opalenik, S.R.; Loi, S.; Zhou, J.; et al. Changes in Peripheral and Local Tumor Immunity after Neoadjuvant Chemotherapy Reshape Clinical Outcomes in Patients with Breast Cancer. Clin. Cancer Res. 2020, 26, 5668–5681. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Vandenberg, C.J.; Lieschke, E.; di Rago, L.; Scott, C.L.; Majewski, I.J. CHK2 Inhibition Provides a Strategy to Suppress Hematologic Toxicity from PARP Inhibitors. Mol. Cancer Res. 2021, 19, 1350–1360. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Molin, G.Z.D.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Lama, L.; Galindo-Campos, M.A.; Martínez, C.; Comerma, L.; Vazquez, I.; Vernet-Tomas, M.; Ampurdanés, C.; Lutfi, N.; Martin-Caballero, J.; Dantzer, F.; et al. Coordinated signals from PARP-1 and PARP-2 are required to establish a proper T cell immune response to breast tumors in mice. Oncogene 2020, 39, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.-C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Luo, H.; Su, J.; Di, S.; Zhou, M.; Shi, B.; Sun, Y.; Du, G.; Zhang, H.; Jiang, H.; et al. Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol. Ther. 2021, 29, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Baldwin, P.; Leal, A.S.M.; Carapellucci, S.; Sridhar, S.; Liby, K.T. A nano-liposome formulation of the PARP inhibitor Talazoparib enhances treatment efficacy and modulates immune cell populations in mammary tumors of BRCA-deficient mice. Theranostics 2019, 9, 6224–6238. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Ghonim, M.A.; Ibba, S.V.; Tarhuni, A.F.; Errami, Y.; Luu, H.H.; Dean, M.J.; El-Bahrawy, A.H.; Wyczechowska, D.; Benslimane, I.A.; del Valle, L.; et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J. Immunother. Cancer 2021, 9, e001643. [Google Scholar] [CrossRef]
- Sen, T.; della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Kadel, S.; Townsend, M.G.; Oliwa, M.; Guerriero, J.L. Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front. Immunol. 2021, 12, 643771. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A.; Martinez-Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Allouch, A.; Paoletti, A.; Leteur, C.; Mirjolet, C.; Martins, I.; Voisin, L.; Law, F.; Dakhli, H.; Mintet, E.; et al. NOX2-dependent ATM kinase activation dictates pro-inflammatory macrophage phenotype and improves effectiveness to radiation therapy. Cell Death Differ. 2017, 24, 1632–1644. [Google Scholar] [CrossRef]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.-R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef]
- Glasner, A.; Plitas, G. Tumor resident regulatory T cells. Semin. Immunol. 2021, 52, 101476. [Google Scholar] [CrossRef] [PubMed]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ Regulatory T Cells in Poly(ADP-Ribose) Polymerase-1 Deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef]
- Navarro, J.; Gozalbo-López, B.; Méndez, A.C.; Dantzer, F.; Schreiber, V.; Martínez, C.; Arana, D.M.; Farrés, J.; Revilla-Nuin, B.; Bueno, M.F.; et al. PARP-1/PARP-2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci. Rep. 2017, 7, 41962. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Campos, M.A.; Lutfi, N.; Bonnin, S.; Martínez, C.; Velasco-Hernandez, T.; García-Hernández, V.; Martin-Caballero, J.; Ampurdanés, C.; Gimeno, R.; Colomo, L.; et al. Distinct roles for PARP-1 and PARP-2 in c-Myc-driven B-cell lymphoma in mice. Blood 2021. [Google Scholar] [CrossRef]
- Ahmad, A.; de Camargo Vieira, J.; de Mello, A.H.; de Lima, T.M.; Ariga, S.K.; Barbeiro, D.F.; Barbeiro, H.V.; Szczesny, B.; Törö, G.; Druzhyna, N.; et al. The PARP inhibitor olaparib exerts beneficial effects in mice subjected to cecal ligature and puncture and in cells subjected to oxidative stress without impairing DNA integrity: A potential opportunity for repurposing a clinically used oncological drug for the experimental therapy of sepsis. Pharmacol. Res. 2019, 145, 104263. [Google Scholar] [CrossRef] [Green Version]
- Proctor, M.; Gonzalez Cruz, J.L.; Daignault-Mill, S.M.; Veitch, M.; Zeng, B.; Ehmann, A.; Sabdia, M.; Snell, C.; Keane, C.; Dolcetti, R.; et al. Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers 2021, 13, 3733. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; LaLonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000340. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Pardoll, D.M.; Topalian, S.L. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 1998, 10, 588–594. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Echeverria Tirado, L.C.; Ghonim, M.A.; Wang, J.; Al-Khami, A.A.; Wyczechowska, D.; Luu, H.H.; Kim, H.; Sanchez-Pino, M.D.; Yélamos, J.; Yassin, L.M.; et al. PARP-1 Is Critical for Recruitment of Dendritic Cells to the Lung in a Mouse Model of Asthma but Dispensable for Their Differentiation and Function. Mediat. Inflamm. 2019, 2019, 1656484. [Google Scholar] [CrossRef] [Green Version]
- Cerwenka, A.; Lanier, L.L. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 2016, 16, 112–123. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Muntasell, A.; Rojo, F.; Servitja, S.; Rubio-Perez, C.; Cabo, M.; Tamborero, D.; Costa-Garcia, M.; Martínez-Garcia, M.; Menéndez, S.; Vazquez, I.; et al. NK Cell Infiltrates and HLA Class I Expression in Primary HER2+ Breast Cancer Predict and Uncouple Pathological Response and Disease-free Survival. Clin. Cancer Res. 2019, 25, 1535–1545. [Google Scholar] [CrossRef] [Green Version]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non–small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Wang, X.-D.; Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. eLife 2020, 9, 1–47. [Google Scholar] [CrossRef]
- MacKenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nat. Cell Biol. 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Chen, W.; Zhu, H.-J. The immune suppressive function of transforming growth factor-β(TGF-β) in human diseases. Growth Factors 2014, 33, 92–101. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; van Vugt, M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nat. Cell Biol. 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 2014, 5, 5166. [Google Scholar] [CrossRef] [Green Version]
- Paludan, S.R.; Reinert, L.S.; Hornung, V. DNA-stimulated cell death: Implications for host defence, inflammatory diseases and cancer. Nat. Rev. Immunol. 2019, 19, 141–153. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Bruand, M.; Barras, D.; Mina, M.; Ghisoni, E.; Morotti, M.; Lanitis, E.; Fahr, N.; Desbuisson, M.; Grimm, A.; Zhang, H.; et al. Cell-autonomous inflammation of BRCA1-deficient ovarian cancers drives both tumor-intrinsic immunoreactivity and immune resistance via STING. Cell Rep. 2021, 36, 109412. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; de Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Gao, Y.; Zeng, Z.; Luo, Y.; Jiang, X.; Zhang, J.; Li, J.; Gong, Y.; Xie, C. PARP inhibitor niraparib as a radiosensitizer promotes antitumor immunity of radiotherapy in EGFR-mutated non-small cell lung cancer. Clin. Transl. Oncol. 2021, 23, 1–11. [Google Scholar] [CrossRef]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1–SPOP Signaling and Enhances Anti–PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Morel, D.; Eychenne, T.; Colmet-Daage, L.; Bajrami, I.; Dorvault, N.; Garrido, M.; Meisenberg, C.; Lamb, A.; Ngo, C.; et al. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 2021, 81, 2888–2902. [Google Scholar] [CrossRef]
- Wu, X.; Kang, X.; Zhang, X.; Xie, W.; Su, Y.; Liu, X.; Guo, L.; Guo, E.; Li, F.; Hu, D.; et al. WEE1 inhibitor and ataxia telangiectasia and RAD3-related inhibitor trigger stimulator of interferon gene-dependent immune response and enhance tumor treatment efficacy through programmed death-ligand 1 blockade. Cancer Sci. 2021, 112, 4444–4456. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.-Y. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J. Clin. Investig. 2021, 131, e139333. [Google Scholar] [CrossRef]
- Wayne, J.; Brooks, T.; Landras, A.; Massey, A.J. Targeting DNA damage response pathways to activate the STING innate immune signaling pathway in human cancer cells. FEBS J. 2021, 288, 4507–4540. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies that are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.N.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: Implications for tumor immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Miao, Y.R.; Thakkar, K.N.; Qian, J.; Kariolis, M.S.; Huang, W.; Nandagopal, S.; Yang, T.T.C.; Diep, A.N.; Cherf, G.M.; Xu, Y.; et al. Neutralization of PD-L2 is Essential for Overcoming Immune Checkpoint Blockade Resistance in Ovarian Cancer. Clin. Cancer Res. 2021, 27, 4435–4448. [Google Scholar] [CrossRef]
- Awadasseid, A.; Wu, Y.; Zhang, W. Advance investigation on synthetic small-molecule inhibitors targeting PD-1/PD-L1 signaling pathway. Life Sci. 2021, 282, 119813. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Nam, A.-R.; Yoon, J.; Jin, M.-H.; Bang, J.-H.; Oh, K.-S.; Seo, H.-R.; Kim, J.-M.; Kim, T.-Y.; Oh, D.-Y. ATR inhibition amplifies antitumor effects of olaparib in biliary tract cancer. Cancer Lett. 2021, 516, 38–47. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Qin, G.; Wang, X.; Ye, S.; Li, Y.; Chen, M.; Wang, S.; Qin, T.; Zhang, C.; Li, Y.; Long, Q.; et al. NPM1 upregulates the transcription of PD-L1 and suppresses T cell activity in triple-negative breast cancer. Nat. Commun. 2020, 11, 1669. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Li, C.-W.; Lim, S.-O.; Sun, L.; Lai, Y.-J.; Hou, J.; Liu, C.; Chang, C.-W.; Qiu, Y.; Hsu, J.-M.; et al. Deglycosylation of PD-L1 by 2-deoxyglucose reverses PARP inhibitor-induced immunosuppression in triple-negative breast cancer. Am. J. Cancer Res. 2018, 8, 1837–1846. [Google Scholar]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Pusztai, L.; Yau, C.; Wolf, D.M.; Han, H.S.; Du, L.; Wallace, A.M.; String-Reasor, E.; Boughey, J.C.; Chien, A.J.; Elias, A.D.; et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 2021, 39, 989–998.e5. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Thara, E.; Awad, M.M.; Dowlati, A.; Haque, B.; Stinchcombe, T.E.; Dy, G.K.; Spigel, D.R.; Lu, S.; Lyer Singh, N. JASPER: Phase 2 trial of first-line niraparib plus pembrolizumab in patients with advanced non-small cell lung cancer. Cancer 2021. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Färkkilä, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.W.; Koh, B.D.; Zhang, J.-S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) Polymerase Inhibitors Sensitize Cancer Cells to Death Receptor-mediated Apoptosis by Enhancing Death Receptor Expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenerty, K.E.; Padget, M.; Wolfson, B.; Gameiro, S.R.; Su, Z.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Hodge, J.W. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J. Immunother. Cancer 2018, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.R.; Raulet, D.H. NKG2D-Deficient Mice Are Defective in Tumor Surveillance in Models of Spontaneous Malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raulet, D.H.; Guerra, N. Oncogenic stress sensed by the immune system: Role of natural killer cell receptors. Nat. Rev. Immunol. 2009, 9, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.R.; le Bert, N.; Ho, S.S.W.; Shen, Y.J.; Tang, M.L.F.; Xiong, G.M.; Croxford, J.L.; Koo, C.X.; Ishii, K.J.; Akira, S.; et al. RAE1 Ligands for the NKG2D Receptor Are Regulated by STING-Dependent DNA Sensor Pathways in Lymphoma. Cancer Res. 2014, 74, 2193–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.-C.; Bai, Y.; Lu, X.-Z.; Qi, C.-J. Effects and Mechanism of PARP Inhibitor Olaparib on the Expression of NKG2D Ligands in HL-60 Cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 1826–1830. [Google Scholar] [PubMed]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR–dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Morisada, M.; Sun, L.; Moore, E.C.; Padget, M.; Hodge, J.W.; Schlom, J.; Gameiro, S.R.; Allen, C.T. Inhibition of WEE1 kinase and cell cycle checkpoint activation sensitizes head and neck cancers to natural killer cell therapies. J. Immunother. Cancer 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Study Identifier | Study Design | Condition | Target | Drug | Primary Outcome |
|---|---|---|---|---|---|
| NCT03955471 | Single Arm | Ovarian Neoplasms | PARP | Olaparib + TSR-042 (anti-PD-1 mAb) | ORR (5 years) |
| NCT03951415 | Single Arm | Endometrial Neoplasms, Uterine Neoplasms | PARP | Olaparib + Durvalumab (anti-PD-L1 mAb) | PFS (12 weeks) |
| NCT03851614 | Two Arms | Mismatch Repair Proficient Colorectal Cancer, Pancreatic Adenocarcinoma, Leiomyosarcoma | PARP | Olaparib + Durvalumab (anti-PD-L1 mAb) | Changes in genomic and immune biomarkers (3 years) |
| NCT03780608 | Single Arm with 2 Cohorts | Gastric Adenocarcinoma, Malignant Melanoma | ATR | Ceralasertib [AZD6738] + Durvalumab (anti-PD-L1 mAb) | ORR (2 years) |
| NCT03574779 | Single Arm | Ovarian Neoplasms | PARP | Niraparib + TSR-042 (anti-PD-1 mAb) + Bevacizumab (anti-VEGF mAb) | ORR (6 years) |
| NCT03565991 | Single Arm with 2 Cohorts | Advanced Solid Tumours with Defects in BRCA1/BRCA2, Advanced Solid Tumours with Defects in ATM | PARP | Talazoparib + Avelumab (anti-PD-L1 mAb) | OR (2 years) |
| NCT03330405 | Two Arms with 10 Cohorts | Locally Advanced or Metastatic Solid Tumors | PARP | Talazoparib + Avelumab (anti-PD-L1 mAb) | ORR (2 years) |
| NCT03167619 | Two Arms | Triple Negative Breast Cancer | PARP | Olaparib + Durvalumab (anti-PD-L1 mAb) | PFS (12 months) |
| NCT02953457 | Single Arm | Recurrent ovarian, fallopian tube, or primary peritoneal cancer with BRCA1 or BRCA2 genetic mutation | PARP | Olaparib + Durvalumab (anti-PD-L1 mAb) + Tremelimumab (anti-CTLA-4 mAb) | PFS (3 and 6 months) |
| NCT02657889 | Single Arm | Triple Negative Breast Cancer, Ovarian Cancer, Fallopian Tube Cancer, Peritoneal Cancer | PARP | Niraparib + Pembrolizumab (anti-PD-1 mAb) | ORR (40 weeks) |
| NCT02571725 | Single Arm | Ovarian Cancer, Fallopian Tube Cancer, Peritoneal Neoplasms | PARP | Olaparib + Tremelimumab (anti-CTLA-4 mAb) | ORR (2 years) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lutfi, N.; Galindo-Campos, M.A.; Yélamos, J. Impact of DNA Damage Response—Targeted Therapies on the Immune Response to Tumours. Cancers 2021, 13, 6008. https://doi.org/10.3390/cancers13236008
Lutfi N, Galindo-Campos MA, Yélamos J. Impact of DNA Damage Response—Targeted Therapies on the Immune Response to Tumours. Cancers. 2021; 13(23):6008. https://doi.org/10.3390/cancers13236008
Chicago/Turabian StyleLutfi, Nura, Miguel Alejandro Galindo-Campos, and José Yélamos. 2021. "Impact of DNA Damage Response—Targeted Therapies on the Immune Response to Tumours" Cancers 13, no. 23: 6008. https://doi.org/10.3390/cancers13236008
APA StyleLutfi, N., Galindo-Campos, M. A., & Yélamos, J. (2021). Impact of DNA Damage Response—Targeted Therapies on the Immune Response to Tumours. Cancers, 13(23), 6008. https://doi.org/10.3390/cancers13236008