
Epigenetic Dysregulation of KCNK9 Imprinting and Triple-Negative Breast Cancer

 , , and
, , and
Abstract
:Simple Summary
Abstract
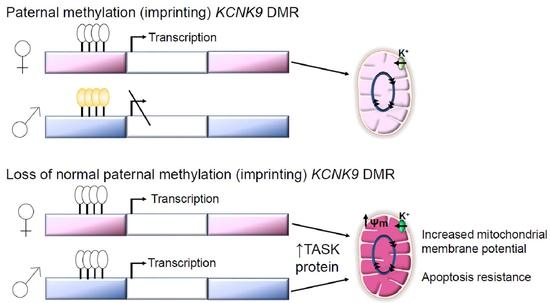
1. Introduction
2. Materials and Methods
3. Results
3.1. KCNK9 Is Imprinted in Mammary Epithelial Cells
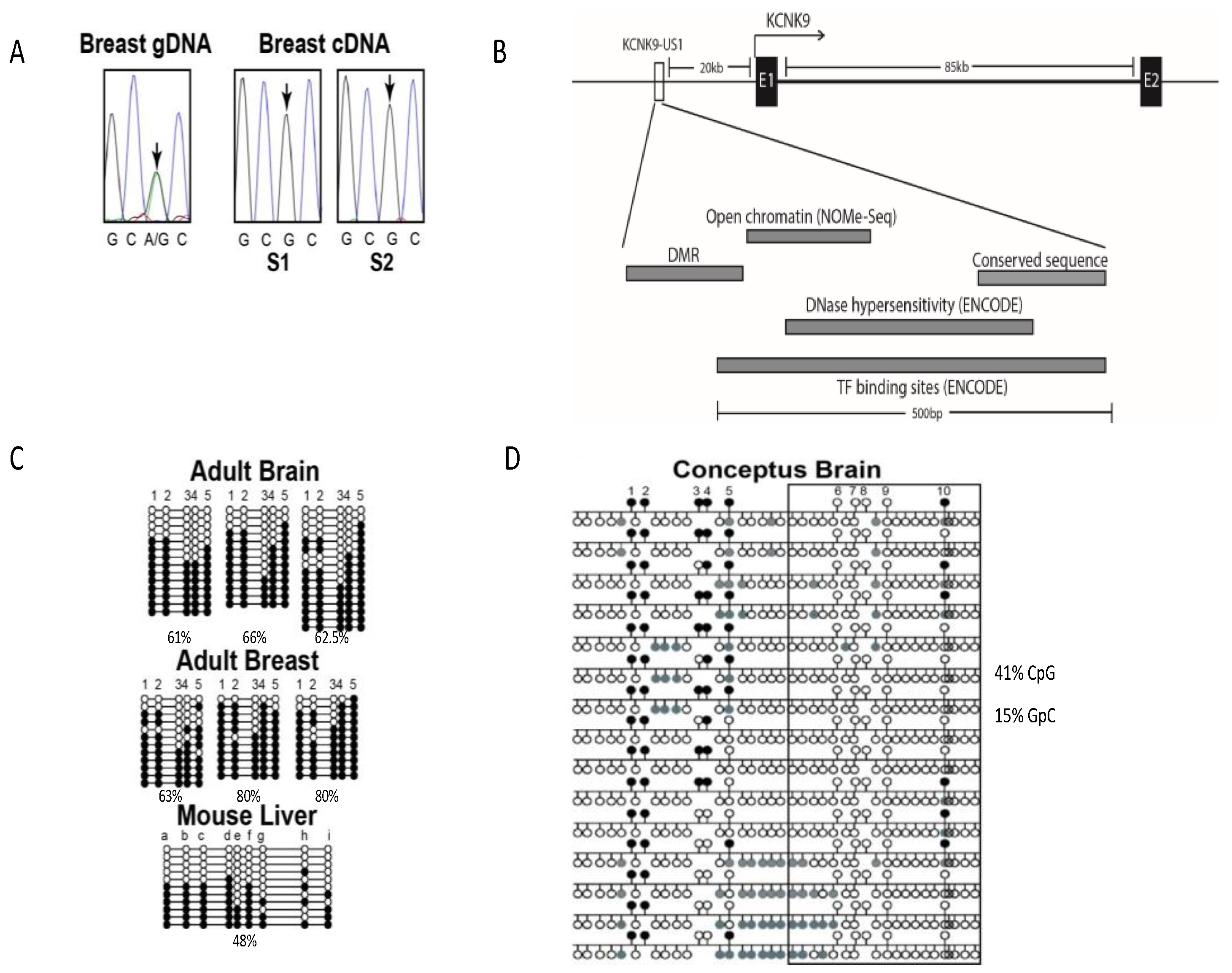
3.1.1. KCNK9 Exhibits Monoallelic Expression in Breast Tissue
3.1.2. Identification of the KCNK9 Imprint DMR
3.1.3. Methylation of the KCNK9 DMR Regulates Chromatin Structure
3.1.4. Methylation Status of the KCNK9 DMR Does Not Correlate with Age
3.2. Hypomethylaiton of the KCNK9 DMR Is Observed in Invasive Breast Cancer; Hypomethylation of the KCNK9 DMR Increases TASK3 Protein Expression
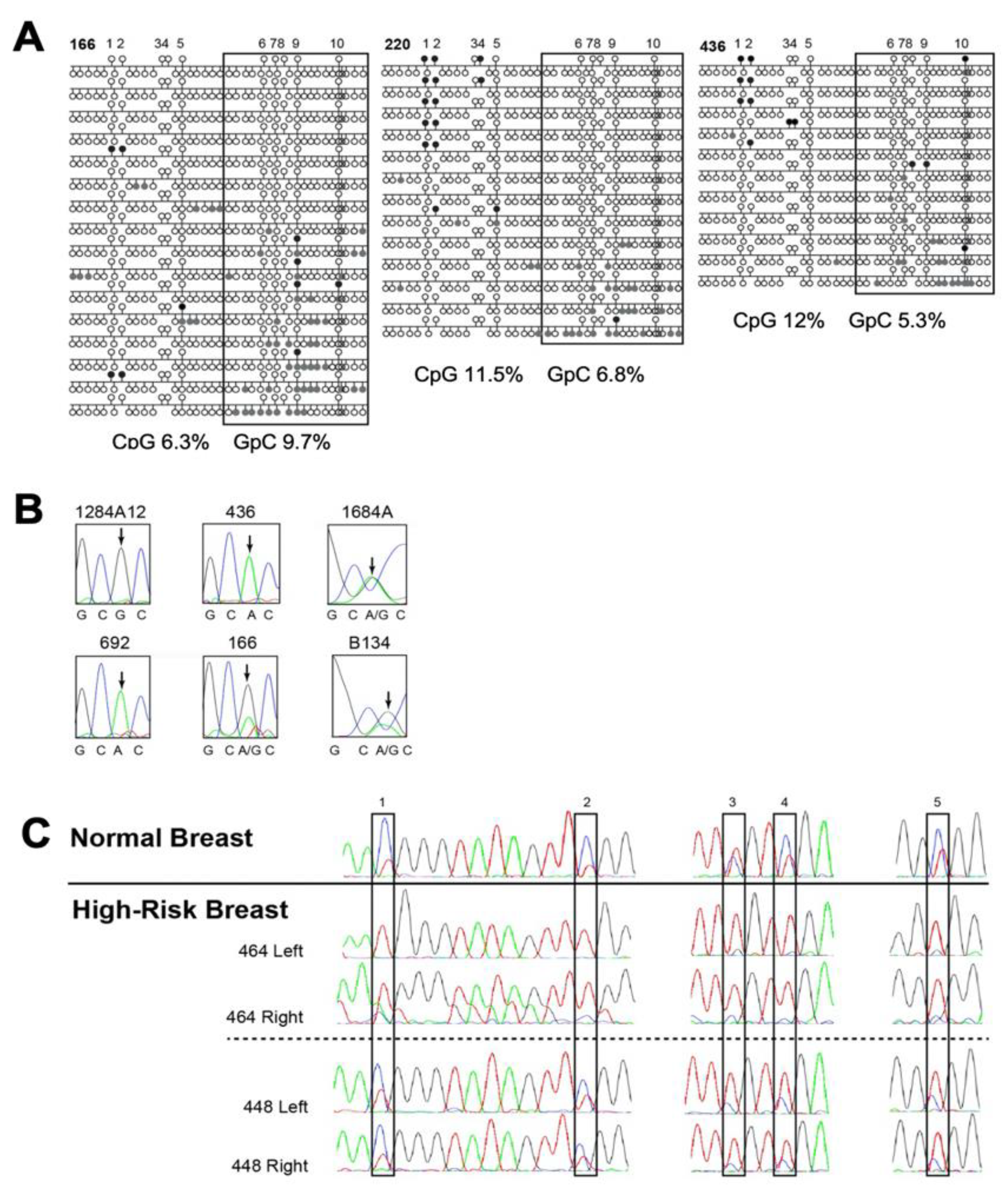
3.2.1. KCNK9 DMR Analysis and Invasive Breast Cancer
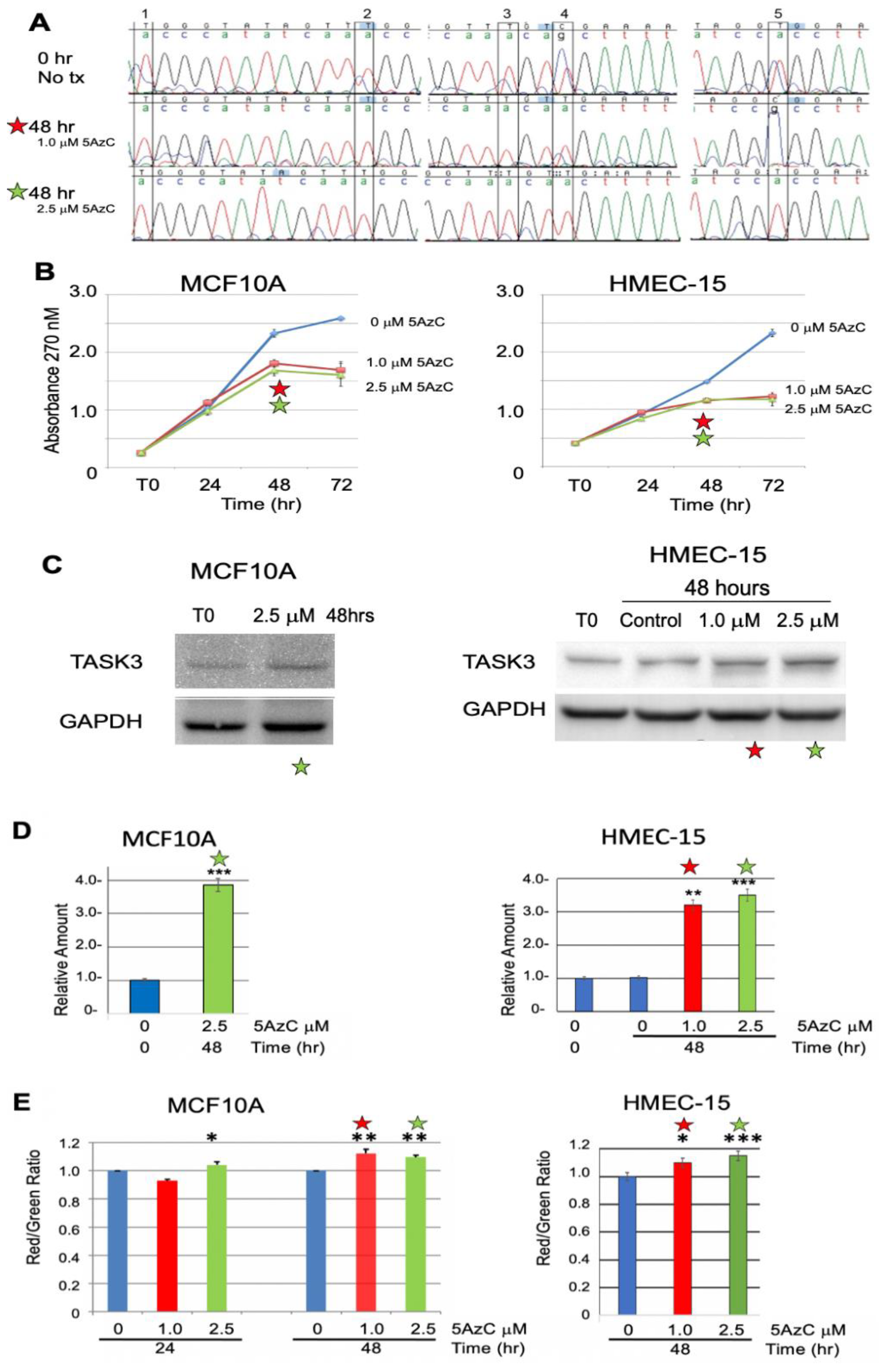
3.2.2. Chemical Demethylation of the KCNK9 DMR Increases Expression of the KCNK9 Gene Product, TASK3
3.3. Exogenous Expression of TASK3 Increases Mitochondrial Expression of TASK3, ΔΨM, and Promotes Resistance to Staurosporine-Induced Apopotosis
3.4. Hypomethylation of the KCNK9 DMR Is Most Frequently Observed in African-American Women with TNBC versus Caucasians with TNBC
3.4.1. High-Risk Sample Set
3.4.2. Hypomethylation of the KCNK9 DMR Was Most Frequently Observed TNBC
3.4.3. KCNK9 DMR Hypomethylation Is Observed in Our Dataset More Frequently in High-Risk African-American Women with TNBC
3.5. Hypomethylation of the KCNK9 DMR Is Observed in Both TNBC and Non-Cancerous Breast Tissue, but Not in WBCs
Hypomethylation of the KCNK9 DMR Methylation Is Observed in Non-Cancerous Tissue
3.6. KCNK9 DMR Hypomethylation Is Not Associated with the Degree of Cytologic Abnormality, but Is Associated with Increased Mitochondrial Membrane Potential
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Materials and Methods
- KCNK9-US1f: 5′-AGGAAGAGAGATATTTAAATGGTAAGGGTAGAG-3′;
- KCNK9-US1r:5′-CAGTAATACGACTCACTATAGGGAGAAGGCTAACCCCCAAAAAACTAAT- AA-3′;
- KCNK9-US2f: 5′-AGGAAGAGAGGGTTTGGGTTAAGATTTTTAAGAT-3′;
- KCNK9-US2r:5′-CAGTAATACGACTCACTATAGGGAGAAGGCTAAAAATTCTTCCTTCCTTC- CA-3′;
- KCNK9-US0: 5′-TATGGGAAAGGCTAAGGGAA-3′;
- KCNK9-US4: 5′-AAATTCTTCCTTCCTTCCACCTTTA-3′.
- TM1R: 5′-ATGCAGTCGCTATAGCCTGACATGGTCAAGAACCTGAGGAC-3′;
- KCDX-1F: 5′-CCTTCTACTTTGCGATCACGGTCATCACCAC-3′;
- KCDX-3F: 5′-TCATCACCACCATAGGTTATGGGCACGCTGCACC-3′;
- KCDX-3R: 5′-CCAGGATATACATAAAGCTAAAGGCCACGTAGAGC-3′;
- KC-S2: 5′-CAAGGCCTTCTGCATGTTCTA-3′.
- F: 5′-GGATCCGCCATGAAGAGGCAGAACGTG-3′
- R: 5′-GGATCCCACGGACGCCTCTCTTGAATG-3′.
- F: 5′-CACCACCATAGAGTATGGGCACGC-3′
- R: 5′-GCGTGCCCATACTCTATGGTGGTG-3′.
- KCNK9-F: 5′-GCTCCTTCTACTTTGCGATCACG-3′;
- KCNK9-R: 5′-CTGGAACATGACCAGTGTCAGC-3′;
- 18S rRNA-F: 5′-GTAACCCGTTGAACCCCATT-3′;
- 18S rRNA-R: 5′-CCATCCAATCGGTAGTAGCG-3′.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KCNK9 Promoter TF-Binding Site | Sample | C Kd | C Lv | C Br | Ad Brst | Ad Lv | Ad Br |
|---|---|---|---|---|---|---|---|
| Chr8:140717586-140718423 | CpG sites | 35 | 37 | 37 | 35 | 37 | 37 |
| Forward – ATTTAGGTGACACTATAGAAATTTTAGTTAAGGAAGGGATGGAGA | Mean | 0.069 | 0.093 | 0.08 | 0.062 | 0.074 | 0.058 |
| Reverse – CAGTAATACGACTCACTATAGGGAGAAGGCTCATCTCAAAAATCCTTCCAATACTC | SD | 0.044 | 0.054 | 0.078 | 0.064 | 0.10 | 0.059 |
| Median | 0.06 | 0.09 | 0.05 | 0.04 | 0.04 | 0.04 | |
| Range | 0–0.15 | 0–0.23 | 0–0.32 | 0–0.34 | 0–0.58 | 0–0.29 | |
| KCNK9 CpG island Promoter-Exon 1 | Sample | C Kd | C Lv | C Br | Ad Brst | Ad Lv | Ad Br |
| Chr8:140715033-140715433 | CpG sites | 4 | 4 | ND | ND | ND | ND |
| Reverse – CAGTAATACGACTCACTATAGGGAGAAGGCTTACAAAATCACCAACTCCAACTACC | SD | 0.012 | 0.052 | ND | ND | ND | ND |
| Median | 0.005 | 0 | ND | ND | ND | ND | |
| Range | 0–0.03 | 0–0.11 | ND | ND | ND | ND | |
| KCNK9 CpG island Exon 1-Intron 1 | Sample | C Kd | C Lv | C Br | Ad Brst | Ad Lv | Ad Br |
| Chr 8: 140714363-140714967 | CpG sites | 14 | 14 | 14 | ND | ND | ND |
| Forward – ATTTAGGTGACACTATAGAAGTTTTAGAATTGGAATTTAGGGGAA | Mean | 0.029 | 0.043 | 0.034 | ND | ND | ND |
| Reverse – CAGTAATACGACTCACTATAGGGAGAAGGCTTCATCACCACCATAAATAAAAACTAAA | SD | 0.021 | 0.054 | 0.036 | ND | ND | ND |
| Median | 0.029 | 0.043 | 0.034 | ND | ND | ND | |
| Range | 0–0.07 | 0–0.2 | 0–0.12 | ND | ND | ND | |
| Reverse – CAGTAATACGACTCACTATAGGGAGAAGGCTTACAAAATCACCAACTCCAACTACC | SD | 0.012 | 0.052 | ND | ND | ND | ND |
| Number | Age (Years) | Race | BRCA Mut. | Mutation | Cancer Subtype | Masood R-MEC | Masood L-MEC | Methylation R-MEC | Methylation L-MEC | Methylation WBC |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 47 | C | ND | ER+ | 13 | 13 | ++ | ++ | + | |
| 2 | 53 | C | ND | NO | 12 | 14 | + | + | ++ | |
| 3 | 39 | C | ND | HER2+ | 20 | 18 | + | +/− | + | |
| 4 | 52 | C | 1 | IVS5-11T > G | TNBC | 15 | 22 | + | − | + |
| 5 | 43 | AA | ND | NO | 16 | 14 | + | − | + | |
| 6 | 38 | C | 1 | M1775R | TNBC | 15 | 12 | +/− | +/− | + |
| 7 | 52 | C | ND | NO | 19 | 15 | − | − | ++ | |
| 8 | 52 | C | ND | NO | 11 | 13 | ND | + | + | |
| 9 | 39 | C | 0 | TNBC | 16 | 15 | + | ND | + | |
| 10 | 34 | H | 2 | 6872del4 | ER+ | 21 | 15 | + | ND | + |
| 11 | 52 | C | ND | TNBC | 16 | ND | +/− | ND | ++ | |
| 12 | 36 | C | ND | HER2+ | 18 | 18 | + | ND | +/− | |
| 13 | 55 | A | 2UV | G2961S | NO | ND | 15 | ND | +/− | +/− |
| 14 | 55 | C | 1 | exon 22 del 510 bp | NO | 14 | 13 | ND | − | +/− |
| 15 | 51 | AA | ND | ER+ | 14 | ND | − | ND | +/− | |
| 16 | 49 | AA | 0 | TNBC | 18 | ND | − | ND | +/− | |
| 17 | 34 | AA | 0 | TNBC | 14 | 23 | ND | − | + | |
| 18 | 43 | AA | 0 | TNBC | 14 | 20 | ND | − | + | |
| 19 | 46 | C | 0 | NO | 9 | 15 | ND | ND | ++ | |
| 20 | 48 | C | ND | ER+ | 16 | ND | ND | ND | ++ | |
| 21 | 50 | C | ND | TNBC | 14 | ND | ND | ND | ++ | |
| 22 | 40 | C | ND | NO | 12 | 13 | ND | ND | + | |
| 23 | 53 | C | ND | NO | 10 | 13 | ND | ND | + | |
| 24 | 51 | C | ND | NO | 9 | 9 | ND | ND | + | |
| 25 | 51 | C | ND | NO | 13 | 11 | ND | ND | + | |
| 26 | 39 | AA | ND | NO | ND | 9 | ND | ND | + | |
| 27 | 49 | AA | ND | HER2+ | 17 | 18 | ND | ND | + | |
| 28 | 47 | AA | ND | HER2+ | 18 | ND | ND | ND | + | |
| 29 | 49 | C | ND | ER+ | 23 | ND | ND | ND | + | |
| 30 | 40 | C | ND | NO | 16 | 15 | ND | ND | + | |
| 31 | 41 | AA | ND | NO | 14 | 13 | ND | ND | + | |
| 32 | 27 | C | 0 | Rad50-R365Q (1094G > A) | TNBC | 15 | ND | ND | ND | +/− |
| 33 | 45 | AA | ND | NO | 13 | ND | ND | ND | − | |
| 34 | 35 | C | ND | ER+ | 15 | 15 | + | ++ | ND | |
| 35 | 42 | C | 0 | NO | 12 | 11 | + | − | ND | |
| 36 | 47 | C | 1 | Missing | NO | 14 | 13 | − | +/− | ND |
| 37 | 51 | C | 1 | Missing | NO | 12 | 14 | − | − | ND |
| 38 | 41 | AA | 0 | TNBC | 16 | 17 | − | − | ND | |
| 39 | 47 | AA | 0 | TNBC | 15 | 19 | − | − | ND | |
| 40 | 42 | AA | ND | TNBC | 18 | 16 | − | − | ND | |
| 41 | 45 | C | ND | TNBC | 19 | 19 | + | ND | ||
| 42 | 36 | C | ND | TNBC | 16 | ND | +/− | ND | ND | |
| 43 | 42 | AA | ND | ER+ | 16 | 20 | + | +/− | ND | |
| 44 | 49 | C | ND | ER+ | 16 | 20 | − | ND | ND | |
| 45 | 53 | C | 0 | ER+ | 16 | 23 | − | ND | ND |
References
- Jirtle, R.L. How Genes and Environment Interact. In Environmental Epigenomics in Health and Disease: Epigenetics and Disease Origins; Jirtle, R.L., Tyson, F.L., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–30. [Google Scholar]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Hoyo, C. Sculpting Our Future: Environmental Nudging of the Imprintome. In Environmental Epigenomics in Health and Disease: Epigenetics and Disease Origins; Jirtle, R.L., Tyson, F.L., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–30. [Google Scholar]
- Das, R.; Hampton, D.D.; Jirtle, R.L. Imprinting evolution and human health. Mamm. Genome 2009, 20, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Moore, G.E. The role of imprinted genes in humans. Mol. Aspects Med. 2013, 34, 826–840. [Google Scholar] [CrossRef]
- Soubry, A.; Hoyo, C.; Jirtle, R.L.; Murphy, S.K. A paternal environmental legacy: Evidence for epigenetic inheritance through the male germ line. Bioessays 2014, 36, 359–371. [Google Scholar] [CrossRef]
- Murphy, S.K.; Huang, Z.; Hoyo, C. Differentially methylated regions of imprinted genes in prenatal, perinatal and postnatal human tissues. PLoS ONE 2012, 7, e40924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyo, C.; Daltveit, A.K.; Iversen, E.; Benjamin-Neelon, S.E.; Fuemmeler, B.; Schildkraut, J.; Murtha, A.P.; Overcash, F.; Vidal, A.C.; Wang, F.; et al. Erythrocyte folate concentrations, CpG methylation at genomically imprinted domains, and birth weight in a multiethnic newborn cohort. Epigenetics 2014, 9, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.C.; Benjamin Neelon, S.E.; Liu, Y.; Tuli, A.M.; Fuemmeler, B.F.; Hoyo, C.; Murtha, A.P.; Huang, Z.; Schildkraut, J.; Overcash, F.; et al. Maternal stress, preterm birth, and DNA methylation at imprint regulatory sequences in humans. Genet. Epigenet. 2014, 6, 37–44. [Google Scholar] [CrossRef]
- Plass, C.; Soloway, P.D. DNA methylation, imprinting and cancer. Eur. J. Hum. Genet. 2002, 10, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Cui, H.; Ohlsson, R. DNA methylation and genomic imprinting: Insights from cancer into epigenetic mechanisms. Semin. Cancer Biol. 2002, 12, 389–398. [Google Scholar] [CrossRef]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; Le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Small, K.S.; Hedman, A.K.; Grundberg, E.; Nica, A.C.; Thorleifsson, G.; Kong, A.; Thorsteindottir, U.; Shin, S.Y.; Richards, H.B.; Soranzo, N.; et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011, 43, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Jirtle, R.L. Epigenome: The program for human health and disease. Epigenomics 2009, 1, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Szyf, M. The early life environment and the epigenome. Biochim. Biophys. Acta 2009, 1790, 878–885. [Google Scholar] [CrossRef]
- Ho, S.M.; Johnson, A.; Tarapore, P.; Janakiram, V.; Zhang, X.; Leung, Y.K. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 2012, 53, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Luedi, P.P.; Dietrich, F.S.; Weidman, J.R.; Bosko, J.M.; Jirtle, R.L.; Hartemink, A.J. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007, 17, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Ruf, N.; Bähring, S.; Galetzka, D.; Pliushch, G.; Luft, F.C.; Nürnberg, P.; Haaf, T.; Kelsey, G.; Zechner, U. Sequence-based bioinformatic prediction and QUASEP identify genomic imprinting of the KCNK9 potassium channel gene in mouse and human. Hum. Mol. Genet. 2007, 16, 2591–2599. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.J.; Lazdunski, M. The 2P-domain K+ channels: Role in apoptosis and tumorigenesis. Pflug. Arch. 2004, 448, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Chen, L.; Zhang, X.; See, L.H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Barel, O.; Shalev, S.A.; Ofir, R.; Cohen, A.; Zlotogora, J.; Shorer, Z.; Mazor, G.; Finer, G.; Khateeb, S.; Zilberberg, N.; et al. Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am. J. Hum. Genet. 2008, 83, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, L.; Wiser, O.; Slavin, A.; Mu, D.; Powers, S.; Jan, L.Y.; Hoey, T. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc. Natl. Acad. Sci. USA 2003, 100, 7803–7807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamaa, A.; Jackson, L.; Asher, V.; Van Shalkwyk, G.; Warren, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and prognostic significance of the oncogenic K2P potassium channel KCNK9 (TASK-3) in ovarian carcinoma. Anticancer Res. 2013, 33, 1401–1408. [Google Scholar] [PubMed]
- Rusznak, Z.; Bakondi, G.; Kosztka, L.; Pocsai, K.; Dienes, B.; Fodor, J.; Telek, A.; Gonczi, M.; Szucs, G.; Csernoch, L. Mitochondrial expression of the two-pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Arch. 2008, 452, 415–426. [Google Scholar] [CrossRef]
- Kosztka, L.; Rusznak, Z.; Nagy, D.; Nagy, Z.; Fodor, J.; Szucs, G.; Telek, A.; Gonczi, M.; Ruzsnavszky, O.; Szentandrassy, N.; et al. Inhibition of TASK-3 (KCNK9) channel biosynthesis changes cell morphology and decreases both DNA content and mitochondrial function of melanoma cells maintained in cell culture. Melanoma Res. 2011, 21, 308–322. [Google Scholar] [CrossRef]
- Nagy, D.; Gonczi, M.; Dienes, B.; Szoor, A.; Fodor, J.; Nagy, Z.; Toth, A.; Fodor, T.; Bai, P.; Szucs, G.; et al. Silencing the KCNK9 potassium channel (TASK-3) gene disturbs mitochondrial function, causes mitochondrial depolarization, and induces apoptosis of human melanoma cells. Arch. Dermatol. Res. 2014, 306, 885–902. [Google Scholar] [CrossRef]
- Bachmann, M.; Rossa, A.; Antoniazzi, G.; Biasutto, L.; Carrer, A.; Campagnaro, M.; Leanza, L.; Gonczi, M.; Csernoch, L.; Paradisi, C.; et al. Synthesis and cellular effects of a mitochondria-targeted inhibitor of the two-pore potassium channel TASK-3. Pharmacol. Res. 2021, 164, 105326. [Google Scholar] [CrossRef]
- Bean, G.R.; Scott, V.; Yee, L.; Ratliff-Daniel, B.; Troch, M.M.; Seo, P.; Bowie, M.L.; Marcom, P.K.; Slade, J.; Kimler, B.F.; et al. Retinoic acid receptor-beta2 promoter methylation in random periareolar fine needle aspiration. Cancer Epidemiol. Prev. Biomark. 2005, 14, 790–798. [Google Scholar] [CrossRef] [Green Version]
- You, J.S.; Kelly, T.K.; De Carvalho, D.D.; Taberlay, P.C.; Liang, G.; Jones, P.A. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc. Natl. Acad. Sci. USA 2011, 108, 14497–14502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietze, E.C.; Caldwell, L.E.; Grupin, S.L.; Mancini, M.; Seewaldt, V.L. Tamoxifen but not 4-hydroxytamoxifen initiates apoptosis in p53(-) normal human mammary epithelial cells by inducing mitochondrial depolarization. J. Biol. Chem. 2001, 276, 5384–5394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seewaldt, V.L.; Kim, J.H.; Caldwell, L.E.; Johnson, B.S.; Swisshelm, K.; Collins, S.J. All-trans-retinoic acid mediates G(1) arrest but not apoptosis of normal human mammary epithelial cells. Cell Growth Differ. 1997, 8, 631–641. [Google Scholar] [PubMed]
- Ibarra-Drendall, C.; Wilke, L.G.; Zalles, C.; Scott, V.; Archer, L.E.; Lem, S.; Yee, L.D.; Lester, J.; Kulkarni, S.; Murekeyisoni, C.; et al. Reproducibility of random periareolar fine needle aspiration in a multi-institutional Cancer and Leukemia Group B (CALGB) cross-sectional study. Cancer Epidemiol. Prev. Biomark. 2009, 18, 1379–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.S.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. ENCODE data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2013, 41, D56–D63. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [Green Version]
- Greenup, R.; Buchanan, A.; Lorizio, W.; Rhoads, K.; Chan, S.; Leedom, T.; King, R.; McLennan, J.; Crawford, B.; Kelly Marcom, P.; et al. Prevalence of BRCA mutations among women with triple-negative breast cancer (TNBC) in a genetic counseling cohort. Ann. Surg. Oncol. 2013, 20, 3254–3258. [Google Scholar] [CrossRef]
- Newman, L.A. Breast cancer disparities: High-risk breast cancer and African ancestry. Surg. Oncol. Clin. 2014, 23, 579–592. [Google Scholar] [CrossRef]
- Court, F.; Camprubi, C.; Garcia, C.V.; Guillaumet-Adkins, A.; Sparago, A.; Seruggia, D.; Sandoval, J.; Esteller, M.; Martin-Trujillo, A.; Riccio, A.; et al. The PEG13-DMR and brain-specific enhancers dictate imprinted expression within the 8q24 intellectual disability risk locus. Epigenet. Chromatin 2014, 7, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista, D.M.; Sigal, Y.M.; Milstein, A.D.; Garrison, J.L.; Zorn, J.A.; Tsuruda, P.R.; Nicoll, R.A.; Julius, D. Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat. Neurosci. 2008, 11, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Luo, L.Q.; Lal, B.; Ma, X.R.; Chen, L.P.; Hann, C.L.; Fulton, A.M.; Leahy, D.J.; Laterra, J.; Li, M. A monoclonal antibody against KCNK9 K+ channel extracellular domain inhibits tumour growth and metastasis. Nat. Commun. 2016, 7, 10339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampfer, M.; Hallowes, R.C.; Hackett, A.J. Growth of normal human mammary cells in culture. In Vitro 1980, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, S.R.; Martens, T.W.; Sattler, C.A.; Gould, M.N. Association of decreased intercellular communication with the immortal but not the tumorigenic phenotype in human mammary epithelial cells. Cancer Res. 1989, 49, 4326–4331. [Google Scholar] [PubMed]




| Individual—Tissue Type | CpG Site 1 | CpG Site 2 | CpG Sites 3 and 4 | CpG Site 5 |
|---|---|---|---|---|
| Ind1—Brain | 0.66 | 0.7 | 0.32 | 0.34 |
| Ind2—Brain | 0.68 | 0.84 | 0.34 | 0.45 |
| Ind3—Breast | 0.58 | 0.57 | 0.33 | 0.35 |
| Ind4—Breast | 0.3 | 0.5 | 0.23 | 0.2 |
| Ind5—Breast | 0.55 | 0.66 | 0.27 | 0.3 |
| Ind6—Liver | 0.42 | 0.48 | 0.57 | 0.5 |
| Ind7—Liver | 0.41 | 0.46 | 0.48 | 0.34 |
| Ind8—Testis | 0.67 | 0.74 | 0.5 | 0.44 |
| Patient Number | Age (Years) | Race | BRCA Mutation | Cancer Subtype | Methylation MEC | Methylation WBC |
|---|---|---|---|---|---|---|
| 1 | 39 | C | 0 | TNBC | + | + |
| 2 | 41 | AA | 0 | TNBC | − | ND |
| 3 | 34 | AA | 0 | TNBC | − | + |
| 4 | 47 | AA | 0 | TNBC | − | ND |
| 5 | 42 | AA | ND | TNBC | − | ND |
| 6 | 52 | C | 1 | TNBC | − | + |
| 7 | 38 | AA | 1 | TNBC | − | + |
| 8 | 49 | C | ND | TNBC | − | ND |
| 9 | 52 | C | ND | TNBC | − | + |
| 10 | 52 | AA | 0 | TNBC | + | ND |
| 11 | 43 | AA | 0 | TNBC | − | + |
| 12 | 49 | C | ND | TNBC | + | ND |
| 13 | 42 | AA | ND | TNBC | − | ND |
| 14 | 46 | C | ND | TNBC | +/− | ND |
| 15 | 50 | AA | ND | TNBC | − | ND |
| 16 | 50 | AA | ND | TNBC | − | ND |
| 17 | 38 | C | 1 | TNBC | +/− | + |
| 18 | 39 | C | 0 | TNBC | + | + |
| 19 | 55 | C | ND | TNBC | +/− | ++ |
| 20 | 41 | AA | 0 | TNBC | − | ND |
| 21 | 47 | AA | 0 | TNBC | − | ND |
| 22 | 42 | AA | ND | TNBC | − | ND |
| 23 | 45 | C | 0 | TNBC | + | ND |
| 24 | 36 | C | 0 | TNBC | +/− | ND |
| 25 | 36 | C | ND | HER2+ | + | +/− |
| 26 | 40 | C | ND | HER2+ | + | + |
| 27 | 42 | C | 0 | HER2+ | − | + |
| 28 | 49 | AA | 0 | HER2+ | + | + |
| 29 | 42 | AA | ND | HER2+ | + | ND |
| 30 | 50 | C | ND | HER2+ | +/− | ND |
| 31 | 53 | AA | ND | HER2+ | +/− | ND |
| 32 | 39 | C | ND | HER2+ | + | + |
| 33 | 36 | C | ND | HER2+ | + | +/− |
| 34 | 49 | AA | ND | HER2+ | − | ND |
| 35 | 47 | AA | ND | HER2+ | − | ND |
| 36 | 47 | C | ND | ER+ | ++ | + |
| 37 | 35 | L/C | 2 | ER+ | + | + |
| 38 | 52 | C | ND | ER+ | − | ++ |
| 39 | 34 | AA | ND | ER+ | ++ | + |
| 40 | 42 | AA | ND | ER+ | + | ND |
| 41 | 51 | AA | ND | ER+ | − | +/− |
| 42 | 48 | C | ND | ER+ | ++ | ND |
| 43 | 35 | C | 0 | ER+ | + | + |
| 44 | 48 | C | ND | ER+ | − | ND |
| 45 | 36 | C | ND | ER+ | − | ND |
| 46 | 28 | C | ND | ER+ | + | ND |
| 47 | 50 | AA | ND | ER+ | + | ND |
| 48 | 53 | C | ND | ER+ | − | ND |
| 49 | 47 | C | ND | ER+ | ++ | + |
| 50 | 35 | C | ND | ER+ | ++ | ND |
| 51 | 42 | AA | ND | ER+ | + | ND |
| 52 | 49 | C | ND | ER+ | − | ND |
| 53 | 53 | C | ND | ER+ | − | ND |
| # | Age (Years) | Race | BRCA Mt | Mutation | Cancer Subtype | Masood R-MEC | Masood L-MEC | Methylation R-MEC | Methylation L-MEC | Methylation WBC |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 47 | C | ND | ER+ | 13 | 13 | ++ | ++ | + | |
| 2 | 53 | C | ND | NO | 12 | 14 | + | + | ++ | |
| 3 | 39 | C | ND | HER2+ | 20 | 18 | + | +/− | + | |
| 4 | 52 | C | 1 | IVS5-11T > G | TNBC | 15 | 22 | + | − | + |
| 5 | 43 | AA | ND | NO | 16 | 14 | + | − | + | |
| 6 | 38 | C | 1 | M1775R | TNBC | 15 | 12 | +/− | +/− | + |
| 7 | 52 | C | ND | NO | 19 | 15 | − | − | ++ | |
| 8 | 52 | C | ND | NO | 11 | 13 | ND | + | + | |
| 9 | 39 | C | 0 | TNBC | 16 | 15 | + | ND | + | |
| 10 | 34 | L/C | 2 | 6872del4 | ER+ | 21 | 15 | + | ND | + |
| 11 | 52 | C | ND | TNBC | 16 | ND | +/− | ND | ++ | |
| 12 | 36 | C | ND | HER2+ | 18 | 18 | + | ND | +/− | |
| 13 | 55 | A | 2UV | G2961S | NO | ND | 15 | ND | +/− | +/− |
| 14 | 55 | C | 1 | exon 22 del 510 bp | NO | 14 | 13 | ND | − | +/− |
| 15 | 51 | AA | ND | ER+ | 14 | ND | − | ND | +/− | |
| 16 | 49 | AA | 0 | TNBC | 18 | ND | − | ND | +/− | |
| 17 | 34 | AA | 0 | TNBC | 14 | 23 | ND | − | + | |
| 18 | 43 | AA | 0 | TNBC | 14 | 20 | ND | − | + | |
| 19 | 46 | C | 0 | NO | 9 | 15 | ND | ND | ++ | |
| 20 | 48 | C | ND | ER+ | 16 | ND | ND | ND | ++ | |
| 21 | 50 | C | ND | TNBC | 14 | ND | ND | ND | ++ | |
| 22 | 40 | C | ND | NO | 12 | 13 | ND | ND | + | |
| 23 | 53 | C | ND | NO | 10 | 13 | ND | ND | + | |
| 24 | 51 | C | ND | NO | 9 | 9 | ND | ND | + | |
| 25 | 51 | C | ND | NO | 13 | 11 | ND | ND | + | |
| 26 | 39 | AA | ND | NO | ND | 9 | ND | ND | + | |
| 27 | 49 | AA | ND | HER2+ | 17 | 18 | ND | ND | + | |
| 28 | 47 | AA | ND | HER2+ | 18 | ND | ND | ND | + | |
| 29 | 49 | C | ND | ER+ | 23 | ND | ND | ND | + | |
| 30 | 40 | C | ND | NO | 16 | 15 | ND | ND | + | |
| 31 | 41 | AA | ND | NO | 14 | 13 | ND | ND | + | |
| 32 | 27 | C | 0 | Rad50/R365Q (1094G > A) | TNBC | 15 | ND | ND | ND | +/− |
| 33 | 45 | AA | ND | NO | 13 | ND | ND | ND | − | |
| 34 | 35 | C | ND | ER+ | 15 | 15 | + | ++ | ND | |
| 35 | 42 | C | 0 | NO | 12 | 11 | + | − | ND | |
| 36 | 47 | C | 1 | Missing | NO | 14 | 13 | − | +/− | ND |
| 37 | 51 | C | 1 | Missing | NO | 12 | 14 | − | − | ND |
| 38 | 41 | AA | 0 | TNBC | 16 | 17 | − | − | ND | |
| 39 | 47 | AA | 0 | TNBC | 15 | 19 | − | − | ND | |
| 40 | 42 | AA | ND | TNBC | 18 | 16 | − | − | ND | |
| 41 | 45 | C | ND | TNBC | 19 | 19 | + | ND | ||
| 42 | 36 | C | ND | TNBC | 16 | ND | +/− | ND | ND | |
| 43 | 42 | AA | ND | ER+ | 16 | 20 | + | +/− | ND | |
| 44 | 49 | C | ND | ER+ | 16 | 20 | − | ND | ND | |
| 45 | 53 | C | 0 | ER+ | 16 | 23 | − | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skaar, D.A.; Dietze, E.C.; Alva-Ornelas, J.A.; Ann, D.; Schones, D.E.; Hyslop, T.; Sistrunk, C.; Zalles, C.; Ambrose, A.; Kennedy, K.; et al. Epigenetic Dysregulation of KCNK9 Imprinting and Triple-Negative Breast Cancer. Cancers 2021, 13, 6031. https://doi.org/10.3390/cancers13236031
Skaar DA, Dietze EC, Alva-Ornelas JA, Ann D, Schones DE, Hyslop T, Sistrunk C, Zalles C, Ambrose A, Kennedy K, et al. Epigenetic Dysregulation of KCNK9 Imprinting and Triple-Negative Breast Cancer. Cancers. 2021; 13(23):6031. https://doi.org/10.3390/cancers13236031
Chicago/Turabian StyleSkaar, David A., Eric C. Dietze, Jackelyn A. Alva-Ornelas, David Ann, Dustin E. Schones, Terry Hyslop, Christopher Sistrunk, Carola Zalles, Adrian Ambrose, Kendall Kennedy, and et al. 2021. "Epigenetic Dysregulation of KCNK9 Imprinting and Triple-Negative Breast Cancer" Cancers 13, no. 23: 6031. https://doi.org/10.3390/cancers13236031
APA StyleSkaar, D. A., Dietze, E. C., Alva-Ornelas, J. A., Ann, D., Schones, D. E., Hyslop, T., Sistrunk, C., Zalles, C., Ambrose, A., Kennedy, K., Idassi, O., Miranda Carboni, G., Gould, M. N., Jirtle, R. L., & Seewaldt, V. L. (2021). Epigenetic Dysregulation of KCNK9 Imprinting and Triple-Negative Breast Cancer. Cancers, 13(23), 6031. https://doi.org/10.3390/cancers13236031