
Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Role of Oxidative Stress in Cancer Progression
2.1. Free Radicals and Oxidative Stress—General Information
2.2. Oxidative Stress Biomarkers for the Determination of Oncopathology
2.3. ROS-Induced Pro-Oncogenic Signaling
3. Role of Inflammation in Cancer Progression
4. Natural Compounds and Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lippman, S.M.; Hawk, E.T. Cancer Prevention: From 1727 to Milestones of the Past 100 Years. Cancer Res. 2009, 69, 5269–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartner, L. Chemotherapy for Oral Cancer. Dent. Clin. N. Am. 2018, 62, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ejlertsen, B. Adjuvant chemotherapy in early breast cancer. Dan. Med. J. 2016, 63, B5222. [Google Scholar] [PubMed]
- Boulos, S.; Mazhar, D. The evolving role of chemotherapy in prostate cancer. Future Oncol. 2017, 13, 1091–1095. [Google Scholar] [CrossRef]
- Allen, C.; Her, S.; Jaffray, D.A. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2. [Google Scholar] [CrossRef]
- Vinod, S.K.; Hau, E. Radiotherapy treatment for lung cancer: Current status and future directions. Respirology 2020, 25 (Suppl. S2), 61–71. [Google Scholar] [CrossRef]
- Chua, B.; Jackson, J.E.; Lin, C.; Veness, M.J. Radiotherapy for early non-melanoma skin cancer. Oral Oncol. 2019, 98, 96–101. [Google Scholar] [CrossRef]
- Wyld, L.; Audisio, R.A.; Poston, G.J. The evolution of cancer surgery and future perspectives. Nat. Rev. Clin. Oncol. 2015, 12, 115–124. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Yang, Y.; Wen, Y.; Bedi, C.; Humphris, G. The relationship between cancer patient’s fear of recurrence and chemotherapy: A systematic review and meta-analysis. J. Psychosom. Res. 2017, 98, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moslehi, J.; Zhang, Q.; Moore, K.J. Crosstalk Between the Heart and Cancer: Beyond Drug Toxicity. Circulation 2020, 142, 684–687. [Google Scholar] [CrossRef]
- Prado, C.M.; Antoun, S.; Sawyer, M.B.; Baracos, V.E. Two faces of drug therapy in cancer: Drug-related lean tissue loss and its adverse consequences to survival and toxicity. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 250–254. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019. Available online: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death (accessed on 11 December 2020).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Pramono, A.A.; Rather, G.M.; Herman, H.; Lestari, K.; Bertino, J.R. NAD- and NADPH-Contributing Enzymes as Therapeutic Targets in Cancer: An Overview. Biomolecules 2020, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Codd, A.S.; Kanaseki, T.; Torigo, T.; Tabi, Z. Cancer stem cells as targets for immunotherapy. Immunology 2018, 153, 304–314. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Gao, C. Topoisomerase Inhibitors and Targeted Delivery in Cancer Therapy. Curr. Top. Med. Chem. 2019, 19, 713–729. [Google Scholar] [CrossRef]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Marí-Alexandre, J.; Carcelén, A.P.; Agababyan, C.; Moreno-Manuel, A.; García-Oms, J.; Calabuig-Fariñas, S.; Gilabert-Estellés, J.; Alexandre, M.; Carcelén, P.; Manuel, M.; et al. Interplay Between MicroRNAs and Oxidative Stress in Ovarian Conditions with a Focus on Ovarian Cancer and Endometriosis. Int. J. Mol. Sci. 2019, 20, 5322. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell. Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension-a current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Li, H.; Hong, T.; Zhu, Q.; Wang, S.; Huang, T.; Li, X.; Lian, Q.; Ge, R.-S. Paraquat exposure delays late-stage Leydig cell differentiation in rats during puberty. Environ. Pollut. 2019, 255 Pt 2, 113316. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Duarte, J.A.; Sanchez-Navarro, A.; Remião, F.; Bastos, M.L.; Carvalho, F. Paraquat Poisonings: Mechanisms of Lung Toxicity, Clinical Features, and Treatment. Crit. Rev. Toxicol. 2008, 38, 13–71. [Google Scholar] [CrossRef]
- Bello-Medina, P.C.; Rodriguez-Martinez, E.; Prado-Alcala, R.A.; Rivas-Arancibia, S. Ozone pollution, oxidative stress, synaptic plasticity, and neurodegeneration. Neurologia 2021, in press. [Google Scholar] [CrossRef]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, A.; García-Vicente, R.; Morales, M.L.; Ortiz-Ruiz, A.; Martínez-López, J.; Linares, M. Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies. Antioxidants 2020, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-beta Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Wigner, P.; Grębowski, R.; Bijak, M.; Saluk-Bijak, J.; Szemraj, J. The Interplay between Oxidative Stress, Inflammation and Angiogenesis in Bladder Cancer Development. Int. J. Mol. Sci. 2021, 22, 4483. [Google Scholar] [CrossRef] [PubMed]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Aschner, M.; Nguyen, T.T.; Sinitskii, A.I.; Santamaría, A.; Bornhorst, J.; Ajsuvakova, O.P.; da Rocha, J.B.T.; Skalny, A.V.; Tinkov, A.A. Isolevuglandins (isoLGs) as toxic lipid peroxidation byproducts and their pathogenetic role in human diseases. Free Radic. Biol. Med. 2021, 162, 266–273. [Google Scholar] [CrossRef]
- Gào, X.; Brenner, H.; Holleczek, B.; Cuk, K.; Zhang, Y.; Anusruti, A.; Xuan, Y.; Xu, Y.; Schöttker, B. Urinary 8-isoprostane levels and occurrence of lung, colorectal, prostate, breast and overall cancer: Results from a large, population-based cohort study with 14 years of follow-up. Free Radic. Biol. Med. 2018, 123, 20–26. [Google Scholar] [CrossRef]
- Mahdavi, R.; Faramarzi, E.; Seyedrezazadeh, E.; Mohammad-Zadeh, M.; Pourmoghaddam, M. Evaluation of Oxidative Stress, Antioxidant Status and Serum Vitamin C Levels in Cancer Patients. Biol. Trace Elem. Res. 2009, 130, 1. [Google Scholar] [CrossRef]
- Kiliç, N.; Taslipinar, M.Y.; Guney, Y.; Tekin, E.; Onuk, E. An Investigation into the Serum Thioredoxin, Superoxide Dismutase, Malondialdehyde, and Advanced Oxidation Protein Products in Patients with Breast Cancer. Ann. Surg. Oncol. 2014, 21, 4139–4143. [Google Scholar] [CrossRef]
- Seraj, A.; Khan, S.; Ak, S.; Ka, R.; Dubey, R. Antioxidants and lipid peroxidation status in women with breast cancer. Int. Med. J. Malays. 2015, 14, 1–6. [Google Scholar]
- Gupta, R.K.; Patel, A.K.; Kumari, R.; Chugh, S.; Shrivastav, C.; Mehra, S.; Sharma, A.N. Interactions between oxidative stress, lipid profile and antioxidants in breast cancer: A case control study. Asian Pac. J. Cancer Prev. 2012, 13, 6295–6298. [Google Scholar] [CrossRef] [Green Version]
- Pande, D.; Negi, R.; Khanna, S.; Khanna, R.; Khanna, H.D. Vascular Endothelial Growth Factor Levels in Relation to Oxidative Damage and Antioxidant Status in Patients with Breast Cancer. J. Breast Cancer 2011, 14, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Kangari, P.; Farahany, T.Z.; Golchin, A.; Ebadollahzadeh, S.; Salmaninejad, A.; Mahboob, S.A.; Nourazarian, A. Enzymatic antioxidant and lipid peroxidation evaluation in the newly diagnosed breast cancer patients in Iran. Asian Pac. J. Cancer Prev. 2018, 19, 3511–3515. [Google Scholar] [CrossRef] [Green Version]
- Peddireddy, V.; Siva Prasad, B.; Gundimeda, S.D.; Penagaluru, P.R.; Mundluru, H.P. Assessment of 8-oxo-7, 8-dihydro-2′-deoxyguanosine and malondialdehyde levels as oxidative stress markers and antioxidant status in non-small cell lung cancer. Biomarkers 2012, 17, 261–268. [Google Scholar] [CrossRef]
- Dillioglugil, M.O.; Mekık, H.; Muezzinoglu, B.; Ozkan, T.A.; Demir, C.G.; Dillioglugil, O. Blood and tissue nitric oxide and malondialdehyde are prognostic indicators of localized prostate cancer. Int. Urol. Nephrol. 2012, 44, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Kosova, F.; Temeltaş, G.; Arı, Z.; Lekili, M. Possible relations between oxidative damage and apoptosis in benign prostate hyperplasia and prostate cancer patients. Tumor Biol. 2014, 35, 4295–4299. [Google Scholar] [CrossRef]
- El Far, M.; Abol Enein, H.; Zakaria, A.; El Gedamy, M. Evaluation of nitric oxide and malondialdehyde levels in serum of Egyptian patients with bladder and renal tumors: Potential use as medicinal biomarkers. World J. Pharm. Pharm. Sci. 2014, 3, 87–101. [Google Scholar]
- Gecit, I.; Aslan, M.; Güneş, M.; Pirincci, N.; Esen, R.; Demir, H.; Ceylan, K. Serum prolidase activity, oxidative stress, and nitric oxide levels in patients with bladder cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 739–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marakala, V.; Malathi, M.; Shivashankara, A.R. Lipid peroxidation and antioxidant vitamin status in oral cavity and oropharyngeal cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 5763–5765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batcioglu, K.; Mehmet, N.; Ozturk, I.C.; Yilmaz, M.; Aydogdu, N.; Erguvan, R.; Uyumlu, B.; Genc, M.; Karagözler, A.A. Lipid Peroxidation and Antioxidant Status in Stomach Cancer. Cancer Investig. 2006, 24, 18–21. [Google Scholar] [CrossRef]
- Wang, Y.K.; Chiang, W.C.; Kuo, F.C.; Wu, M.C.; Shih, H.Y.; Wang, S.S.W.; Liu, C.J.; Chen, Y.H.; Wu, D.C.; Su, W.W.; et al. Levels of malondialdehyde in the gastric juice: Its association with Helicobacter pylori infection and stomach diseases. Helicobacter 2018, 23, e12460. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.; Rong, S.; Qu, H.; Zhang, Y.; Chang, D.; Pan, H.; Wang, W. Relation between Gastric Cancer and Protein Oxidation, DNA Damage, and Lipid Peroxidation. Oxidative Med. Cell. Longev. 2013, 2013, 543760. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, S.S.; Ghone, R.A.; Suryakar, A.N.; Hundekar, P.S. Lipid peroxidation and antioxidant vitamin status in colorectal cancer patients. Indian J. Physiol. Pharmacol. 2011, 55, 72–76. [Google Scholar]
- Lei, L.; Zhang, J.; Decker, E.A.; Zhang, G. Roles of Lipid Peroxidation-Derived Electrophiles in Pathogenesis of Colonic Inflammation and Colon Cancer. Front. Cell Dev. Biol. 2021, 9, 665591. [Google Scholar] [CrossRef]
- Bitla, A.R.; Reddy, E.P.; Sambasivaih, K.; Suchitra, M.M.; Reddy, V.S.; Srinivasa Rao, P.V.L.N. Evaluation of plasma malondialdehyde as a biomarker in patients with carcinoma of stomach. Biomed. Res. 2011, 22, 63–68. [Google Scholar]
- Saed, G.M.; Diamond, M.P.; Fletcher, N.M. Updates of the role of oxidative stress in the pathogenesis of ovarian cancer. Gynecol. Oncol. 2017, 145, 595–602. [Google Scholar] [CrossRef]
- Manimaran, A.; Rajneesh, C.P. Activities of antioxidant enzyme and lipid peroxidation in ovarian cancer patients. Acad. J. Cancer Res. 2009, 2, 68–72. [Google Scholar]
- Szymańska, B.; Sawicka, E.; Matuszewski, M.; Dembowski, J.; Piwowar, A. The Dependence between Urinary Levels of Angiogenesis Factors, 8-Iso-prostaglandin F2α, ɣ-Synuclein, and Interleukin-13 in Patients with Bladder Cancer: A Pilot Study. J. Oncol. 2020, 2020, 4848752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Chen, B.; Zhang, J.-J.; Li, J.; Yang, Q.; Zhong, Q.-S.; Zhan, S.; Liu, H.; Cai, C. Serum polyunsaturated fatty acid metabolites as useful tool for screening potential biomarker of colorectal cancer. Prostaglandins, Leukot. Essent. Fat. Acids 2017, 120, 25–31. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Nakamura, T.; Takenouchi, S.; Hayashi, A.; Omori, K.; Murata, T. Urinary 8-iso PGF2alpha and 2,3-dinor-8-iso PGF2alpha can be indexes of colitis-associated colorectal cancer in mice. PLoS ONE 2021, 16, e0245292. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Wang, Y.; Guo, W.; Zhou, Y.; Lv, C.; Chen, X.; Liu, K. The significance of the alteration of 8-OHdG in serous ovarian carcinoma. J. Ovarian Res. 2013, 6, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.D.; Cai, Q.; Shu, X.O.; Nechuta, S.J. The Role of Biomarkers of Oxidative Stress in Breast Cancer Risk and Prognosis: A Systematic Review of the Epidemiologic Literature. J. Women Health 2017, 26, 467–482. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Takeda, H.; Otake, S.; Yokozawa, J.; Nishise, S.; Fujishima, S.; Orii, T.; Fukui, T.; Takano, J.; Sasaki, Y.; et al. Increased Plasma Levels of 8-Hydroxydeoxyguanosine Are Associated with Development of Colorectal Tumors. J. Clin. Biochem. Nutr. 2010, 47, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Srivastava, J.K.; Shankar, E.; Kanwal, R.; Nawab, A.; Sharma, H.; Bhaskaran, N.; Ponsky, L.E.; Fu, P.; MacLennan, G.T.; et al. Oxidative Stress and Antioxidant Status in High-Risk Prostate Cancer Subjects. Diagnostics 2020, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Kubo, N.; Morita, M.; Nakashima, Y.; Kitao, H.; Egashira, A.; Saeki, H.; Oki, E.; Kakeji, Y.; Oda, Y.; Maehara, Y. Oxidative DNA damage in human esophageal cancer: Clinicopathological analysis of 8-hydroxydeoxyguanosine and its repair enzyme. Dis. Esophagus 2014, 27, 285–293. [Google Scholar] [CrossRef]
- Yen, C.-J.; Hung, C.-H.; Tsai, W.-M.; Cheng, H.-C.; Yang, H.-L.; Lu, Y.-J.; Tsai, K.-L. Effect of Exercise Training on Exercise Tolerance and Level of Oxidative Stress for Head and Neck Cancer Patients Following Chemotherapy. Front. Oncol. 2020, 10, 1536. [Google Scholar] [CrossRef]
- Kumar, A.; Pant, M.C.; Singh, H.S.; Khandelwal, S. Determinants of oxidative stress and DNA damage (8-OhdG) in squamous cell carcinoma of head and neck. Indian J. Cancer 2012, 49, 309–315. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.-B.; Li, B.; Zhang, Y.; Zhu, Y.-T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Ohtsuka, J.; Oshima, H.; Ezawa, I.; Abe, R.; Oshima, M.; Ohki, R. Functional loss of p53 cooperates with the in vivo microenvironment to promote malignant progression of gastric cancers. Sci. Rep. 2018, 8, 2291. [Google Scholar] [CrossRef]
- Gurram, B.; Zhang, S.; Li, M.; Li, H.; Xie, Y.; Cui, H.; Du, J.; Fan, J.; Wang, J.; Peng, X. Celecoxib Conjugated Fluorescent Probe for Identification and Discrimination of Cyclooxygenase-2 Enzyme in Cancer Cells. Anal. Chem. 2018, 90, 5187–5193. [Google Scholar] [CrossRef]
- Yue, X.; Nguyen, T.D.; Zellmer, V.; Zhang, S.; Zorlutuna, P. Stromal cell-laden 3D hydrogel microwell arrays as tumor microenvironment model for studying stiffness dependent stromal cell-cancer interactions. Biomaterials 2018, 170, 37–48. [Google Scholar] [CrossRef]
- Sicking, I.; Rommens, K.; Battista, M.J.; Böhm, D.; Gebhard, S.; Lebrecht, A.; Cotarelo, C.; Hoffmann, G.; Hengstler, J.G.; Schmidt, M. Prognostic influence of cyclooxygenase-2 protein and mRNA expression in node-negative breast cancer patients. BMC Cancer 2014, 14, 952. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.S.; O’Brien, E.R.; Andreou, K.; Scrace, S.F.; Zakaria, R.; Jenkinson, M.D.; O’Neill, E.; Sibson, N.R. Disruption of tumour-host communication by downregulation of LFA-1 reduces COX-2 and e-NOS expression and inhibits brain metastasis growth. Oncotarget 2016, 7, 52375–52391. [Google Scholar] [CrossRef] [Green Version]
- Sorski, L.; Melamed, R.; Matzner, P.; Lavon, H.; Shaashua, L.; Rosenne, E.; Ben-Eliyahu, S. Reducing liver metastases of colon cancer in the context of extensive and minor surgeries through beta-adrenoceptors blockade and COX2 inhibition. Brain Behav. Immun. 2016, 58, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höing, B.; Kanaan, O.; Altenhoff, P.; Petri, R.; Thangavelu, K.; Schlüter, A.; Lang, S.; Bankfalvi, A.; Brandau, S. Stromal versus tumoral inflammation differentially contribute to metastasis and poor survival in laryngeal squamous cell carcinoma. Oncotarget 2018, 9, 8415–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameziane-El-Hassani, R.; Schlumberger, M.; Dupuy, C. NADPH oxidases: New actors in thyroid cancer? Nat. Rev. Endocrinol. 2016, 12, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.T.; Gao, Y.J.; Ge, Z.Z. NOX4, a new genetic target for anti-cancer therapy in digestive system cancer. J. Dig. Dis. 2018, 19, 578–585. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef] [PubMed]
- Kielbik, M.; Szulc-Kielbik, I.; Klink, M. The Potential Role of iNOS in Ovarian Cancer Progression and Chemoresistance. Int. J. Mol. Sci. 2019, 20, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, L.; Xie, Z.; Zhou, S.; Li, Y.; Zhou, Y.; Sun, M. Nitric Oxide (NO) and NO Synthases (NOS)-Based Targeted Therapy for Colon Cancer. Cancers 2020, 12, 1881. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Pedrosa, R.C.; Dejeans, N.; Glorieux, C.; Levêque, P.; Gallez, B.; Taper, H.; Eeckhoudt, S.; Knoops, L.; Calderon, P.B.; et al. Ascorbate/menadione-induced oxidative stress kills cancer cells that express normal or mutated forms of the oncogenic protein Bcr-Abl. An in vitro and in vivo mechanistic study. Investig. New Drugs 2011, 29, 891–900. [Google Scholar] [CrossRef]
- Glorieux, C.; Dejeans, N.; Sid, B.; Beck, R.; Calderon, P.B.; Verrax, J. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 2011, 82, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Ruottinen, M.; Kaaronen, V.; Saimanen, I.; Kuosmanen, V.; Kärkkäinen, J.; Selander, T.; Aspinen, S.; Eskelinen, M. The Induction of Antioxidant Catalase Enzyme With Decrease of Plasma Malonidialdehyde: An Important Reactive Oxidative Species Inhibiting Mechanism. Anticancer Res. 2020, 40, 5701–5706. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-N.; Lee, J.M.; Oh, H.; Park, J.-H. Glutathione Peroxidase 3 Inhibits Prostate Tumorigenesis in TRAMP Mice. Prostate 2016, 76, 1387–1398. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, Z.; Yingji, S.; Kim, H.; Jin, R.; Renshu, L.; Lee, D.Y.; Roh, M.R.; Yang, S. Downregulation of glutathione peroxidase 3 is associated with lymph node metastasis and prognosis in cervical cancer. Oncol. Rep. 2014, 31, 2587–2592. [Google Scholar] [CrossRef] [Green Version]
- Shaaban, Y.; Aref, S.; Taalab, M.; Ayed, M.; Mabed, M. Implications of Glutathione Peroxidase 3 Expression in a Cohort of Egyptian Patients with Acute Myeloid Leukemia. Asian Pac. J. Cancer Prev. 2020, 21, 3567–3572. [Google Scholar] [CrossRef]
- Robbins, D.; Zhao, Y. Manganese Superoxide Dismutase in Cancer Prevention. Antioxid. Redox Signal. 2014, 20, 1628–1645. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, Y.; Liu, Q. Expression of superoxide dismutase 2 in breast cancer and its clinical significance. Nan Fang Yi Ke Da Xue Xue Bao 2020, 40, 1103–1111. [Google Scholar]
- Islinger, M.; Li, K.W.; Seitz, J.; Völkl, A.; Lüers, G.H. Hitchhiking of Cu/Zn Superoxide Dismutase to Peroxisomes—Evidence for a Natural Piggyback Import Mechanism in Mammals. Traffic 2009, 10, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Eleutherio, E.C.A.; Magalhães, R.S.S.; de Araujo Brasil, A.; Neto, J.R.M.; de Holanda Paranhos, L. SOD1, more than just an antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701. [Google Scholar] [CrossRef] [PubMed]
- Witte, I.; Altenhöfer, S.; Wilgenbus, P.; Amort, J.; Clement, A.M.; Pautz, A.; Li, H.; Förstermann, U.; Horke, S. Beyond reduction of atherosclerosis: PON2 provides apoptosis resistance and stabilizes tumor cells. Cell Death Dis. 2011, 2, e112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, I.; Foerstermann, U.; Devarajan, A.; Reddy, S.T.; Horke, S. Protectors or Traitors: The Roles of PON2 and PON3 in Atherosclerosis and Cancer. J. Lipids 2012, 2012, 342806. [Google Scholar] [CrossRef] [Green Version]
- Bacchetti, T.; Ferretti, G.; Sahebkar, A. The role of paraoxonase in cancer. Semin. Cancer Biol. 2019, 56, 72–86. [Google Scholar] [CrossRef]
- You, X.; Ma, M.; Hou, G.; Hu, Y.; Shi, X. Gene expression and prognosis of NOX family members in gastric cancer. OncoTargets Ther. 2018, 11, 3065–3074. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.-T.; Lin, X.-L.; Wu, S.; Liang, Q.; Yang, L.; Gao, Y.-J.; Ge, Z.-Z. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway. Cell. Signal. 2018, 46, 52–63. [Google Scholar] [CrossRef]
- Zeng, C.; Wu, Q.; Wang, J.; Yao, B.; Ma, L.; Yang, Z.; Li, J.; Liu, B. NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells. Free Radic. Biol. Med. 2016, 101, 236–248. [Google Scholar] [CrossRef]
- Degasper, C.; Brunner, A.; Sampson, N.; Tsibulak, I.; Wieser, V.; Welponer, H.; Marth, C.; Fiegl, H.; Zeimet, A.G. NADPH oxidase 4 expression in the normal endometrium and in endometrial cancer. Tumor Biol. 2019, 41, 1010428319830002. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.-J.; Huang, Y.-X.; Wang, W.; Zhang, Y.; Liu, B.-J.; Qiu, J.-G.; Jiang, B.-H.; Liu, L.-Z. NOX4 Signaling Mediates Cancer Development and Therapeutic Resistance through HER3 in Ovarian Cancer Cells. Cells 2021, 10, 1647. [Google Scholar] [CrossRef]
- Shimada, K.; Fujii, T.; Anai, S.; Fujimoto, K.; Konishi, N. ROS generation via NOX4 and its utility in the cytological diagnosis of urothelial carcinoma of the urinary bladder. BMC Urol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Strycharz-Dudziak, M.; Fołtyn, S.; Dworzański, J.; Kiełczykowska, M.; Malm, M.; Drop, B.; Polz-Dacewicz, M. Glutathione Peroxidase (GPx) and Superoxide Dismutase (SOD) in Oropharyngeal Cancer Associated with EBV and HPV Coinfection. Viruses 2020, 12, 1008. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lei, J.; He, L.; Fan, X.; Yi, F.; Zhang, W. Evaluation and Monitoring of Superoxide Dismutase (SOD) Activity and its Clinical Significance in Gastric Cancer: A Systematic Review and Meta-Analysis. Med. Sci. Monit. 2019, 25, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Strycharz-Dudziak, M.; Kiełczykowska, M.; Drop, B.; Świątek, Ł.; Kliszczewska, E.; Musik, I.; Polz-Dacewicz, M. Total Antioxidant Status (TAS), Superoxide Dismutase (SOD), and Glutathione Peroxidase (GPx) in Oropharyngeal Cancer Associated with EBV Infection. Oxidative Med. Cell. Longev. 2019, 2019, 5832410. [Google Scholar] [CrossRef] [PubMed]
- Ahmed Amar, S.A.; Eryilmaz, R.; Demir, H.; Aykan, S.; Demir, C. Determination of oxidative stress levels and some antioxidant enzyme activities in prostate cancer. Aging Male 2019, 22, 198–206. [Google Scholar] [CrossRef]
- Moustafa, S.R. Association of Superoxide Dismutase, Glutathione Peroxidase, Catalse, and Xanthine Oxidase with Incidence of Bladder Cancer. Cancer Res. J. 2015, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, E.; Jablonowski, Z.; Tomasik, B.; Gromadzinska, J.; Jablonska, E.; Konecki, T.; Fendler, W.; Sosnowski, M.; Wasowicz, W.; Reszka, E. Different Gene Expression and Activity Pattern of Antioxidant Enzymes in Bladder Cancer. Anticancer Res. 2017, 37, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Caglayan, A.; Katlan, D.C.; Selcuk Tuncer, Z.; Yüce, K.; Sayal, H.B.; Coskun Salman, M.; Kocer-Gumusel, B. Impaired antioxidant enzyme functions with increased lipid peroxidation in epithelial ovarian cancer. IUBMB Life 2017, 69, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Zinczuk, J.; Maciejczyk, M.; Zareba, K.; Romaniuk, W.; Markowski, A.; Kedra, B.; Zalewska, A.; Pryczynicz, A.; Matowicka-Karna, J.; Guzinska-Ustymowicz, K. Antioxidant Barrier, Redox Status, and Oxidative Damage to Biomolecules in Patients with Colorectal Cancer. Can Malondialdehyde and Catalase Be Markers of Colorectal Cancer Advancement? Biomolecules 2019, 9, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobanoglu, U.; Demir, H.; Duran, M.; Şehitogullari, A.; Mergan, D.; Demir, C. Erythrocyte catalase and carbonic anhydrase activities in lung cancer. Asian Pac. J. Cancer Prev. 2010, 11, 1377–1382. [Google Scholar] [PubMed]
- Wang, J.Y.; Wang, X.; Wang, X.J.; Zheng, B.Z.; Wang, Y.; Wang, X.; Liang, B. Curcumin inhibits the growth via Wnt/beta-catenin pathway in non-small-cell lung cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7492–7499. [Google Scholar] [PubMed]
- Wei, R.; Qiu, H.; Xu, J.; Mo, J.; Liu, Y.; Gui, Y.; Huang, G.; Zhang, S.; Yao, H.; Huang, X.; et al. Expression and prognostic potential of GPX1 in human cancers based on data mining. Ann. Transl. Med. 2020, 8, 124. [Google Scholar] [CrossRef]
- Wei, J.; Xie, Q.; Liu, X.; Wan, C.; Wu, W.; Fang, K.; Yao, Y.; Cheng, P.; Deng, D.; Liu, Z. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann. Transl. Med. 2020, 8, 678. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, W.; Jiang, N.; Zhang, L.; Li, Y.; Xu, X.; Cai, S.; Wei, L.; Liu, X.; Chen, G.; et al. Clinical Implications of iNOS Levels in Triple-Negative Breast Cancer Responding to Neoadjuvant Chemotherapy. PLoS ONE 2015, 10, e0130286. [Google Scholar] [CrossRef]
- Ranganathan, S.; Krishnan, A.; Sivasithambaram, N.D. Significance of twist and iNOS expression in human breast carcinoma. Mol. Cell. Biochem. 2016, 412, 41–47. [Google Scholar] [CrossRef]
- Hong, S.K.; Gul, Y.A.; Ithnin, H.; Talib, A.; Seow, H.F. Expression of beta-catenin, COX-2 and iNOS in colorectal cancer: Relevance of COX-2 adn iNOS inhibitors for treatment in Malaysia. Asian J. Surg. 2004, 27, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhou, S.; Pang, L.; Yang, J.; Li, H.J.; Huo, X.; Qian, S.Y. Celastrol suppresses nitric oxide synthases and the angiogenesis pathway in colorectal cancer. Free Radic. Res. 2019, 53, 324–334. [Google Scholar] [CrossRef]
- Sandes, E.O.; Lodillinsky, C.; Langle, Y.; Belgorosky, D.; Marino, L.; Gimenez, L.; Casabe, A.R.; Eijan, A.M. Inducible nitric oxide synthase and PPARgamma are involved in bladder cancer progression. J. Urol. 2012, 188, 967–973. [Google Scholar] [CrossRef]
- Kilic, S.; Bayraktar, N.; Beytur, A.; Ergin, H.; Bayraktar, M.; Egri, M. Can the levels of nitric oxide in the urine, serum and tumor tissue be putative markers for bladder cancer? Int. J. Urol. 2006, 13, 1079–1085. [Google Scholar] [CrossRef]
- Wang, J.; Hussain, S.P. NO• and Pancreatic Cancer: A Complex Interaction with Therapeutic Potential. Antioxid. Redox Signal. 2017, 26, 1000–1008. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Tsolou, A.; Daridou, E.; Kouroupi, M.; Chlichlia, K.; Koukourakis, M.I. iNOS Expression by Tumor-Infiltrating Lymphocytes, PD-L1 and Prognosis in Non-Small-Cell Lung Cancer. Cancers 2020, 12, 3276. [Google Scholar] [CrossRef]
- Celenk, F.; Bayramoglu, I.; Yilmaz, A.; Menevse, A.; Bayazit, Y. Expression of Cyclooxygenase-2, 12-Lipoxygenase, and Inducible Nitric Oxide Synthase in Head and Neck Squamous Cell Carcinoma. J. Craniofac. Surg. 2013, 24, 1114–1117. [Google Scholar] [CrossRef]
- Girotti, A.W.; Fahey, J.M.; Korytowski, W. Nitric oxide-elicited resistance to anti-glioblastoma photodynamic therapy. Cancer Drug Resist. 2020, 3, 401–414. [Google Scholar] [CrossRef]
- Ding, Z.; Ogata, D.; Roszik, J.; Qin, Y.; Kim, S.-H.; Tetzlaff, M.T.; Lazar, A.J.; Davies, M.A.; Ekmekcioglu, S.; Grimm, E.A. iNOS Associates With Poor Survival in Melanoma: A Role for Nitric Oxide in the PI3K-AKT Pathway Stimulation and PTEN S-Nitrosylation. Front. Oncol. 2021, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, M.A.; Cenciarelli, C.; Tesori, V.; Cappellari, M.; Martini, M.; Di Francesco, A.M.; Giorda, E.; Carsetti, R.; Ricci-Vitiani, L.; Gasbarrini, A. High nitric oxide production, secondary to inducible nitric oxide synthase expression, is essential for regulation of the tumour-initiating properties of colon cancer stem cells. J. Pathol. 2015, 236, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Yongsanguanchai, N.; Pongrakhananon, V.; Mutirangura, A.; Rojanasakul, Y.; Chanvorachote, P. Nitric oxide induces cancer stem cell-like phenotypes in human lung cancer cells. Am. J. Physiol. Cell Physiol. 2015, 308, C89–C100. [Google Scholar] [CrossRef] [Green Version]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Gałczyński, K.; Bełtowski, J.; Nowakowski, Ł.; Vasilevska, D.; Rechberger, T.; Semczuk, A. Serum paraoxonase 1 activity and protein N-homocysteinylation in primary human endometrial cancer. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihajlovic, M.; Gojkovic, T.; Vladimirov, S.; Miljkovic, M.; Stefanovic, A.; Vekic, J.; Zeljkovic, D.; Trifunovic, B.; Kotur-Stevuljevic, J.; Spasojevic-Kalimanovska, V.; et al. Changes in lecithin: Cholesterol acyltransferase, cholesteryl ester transfer protein and paraoxonase-1 activities in patients with colorectal cancer. Clin. Biochem. 2019, 63, 32–38. [Google Scholar] [CrossRef]
- Utangac, M.M.; Yeni, E.; Savas, M.; Altunkol, A.; Ciftci, H.; Gumus, K.; Demir, M. Paraoxonase and arylesterase activity in bladder cancer. Türk Üroloji Derg./Turk. J. Urol. 2017, 43, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Iftimie, S.; García-Heredia, A.; Pujol-Bosch, F.; Pont-Salvadó, A.; López-Azcona, A.F.; Hernández-Aguilera, A.; Cabré, N.; Luciano-Mateo, F.; Fort-Gallifa, I.; Castro, A.; et al. Serum Paraoxonase-1 Concentration as a Potential Predictor of Urinary Bladder Cancer Recurrence. A Five Year Follow-Up Study. Arch. Med. Res. 2018, 49, 119–122. [Google Scholar] [CrossRef]
- Bacchetti, T.; Salvolini, E.; Pompei, V.; Campagna, R.; Molinelli, E.; Brisigotti, V.; Togni, L.; Lucarini, G.; Sartini, D.; Campanati, A.; et al. Paraoxonase-2: A potential biomarker for skin cancer aggressiveness. Eur. J. Clin. Investig. 2021, 51, e13452. [Google Scholar] [CrossRef]
- Wang, X.; Xu, G.; Zhang, J.; Wang, S.; Ji, M.; Mo, L.; Zhu, M.; Li, J.; Zhou, G.; Lu, J.; et al. The clinical and prognostic significance of paraoxonase-2 in gastric cancer patients: Immunohistochemical analysis. Hum. Cell 2019, 32, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Bacchetti, T.; Sartini, D.; Pozzi, V.; Cacciamani, T.; Ferretti, G.; Emanuelli, M. Exploring the role of Paraoxonase-2 in bladder cancer: Analyses performed on tissue samples, urines and cell cultures. Oncotarget 2017, 8, 28785–28795. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Pabst, A.M.; Al-Nawas, B.; Horke, S.; Moergel, M. Paraoxonase-2 (PON2) protects oral squamous cell cancer cells against irradiation-induced apoptosis. J. Cancer Res. Clin. Oncol. 2015, 141, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.-H.; Chen, C.-Y.; Chen, P.-C.; Hsiao, S.-H.; Fan, C.-C.; Liang, Y.-C.; Chen, C.-P. Valproic acid inhibits glioblastoma multiforme cell growth via paraoxonase 2 expression. Oncotarget 2017, 8, 14666–14679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Patel, A.K.; Shah, N.; Choudhary, A.K.; Jha, U.K.; Yadav, U.C.; Gupta, P.K.; Pakuwal, U. Oxidative Stress and Antioxidants in Disease and Cancer: A Review. Asian Pac. J. Cancer Prev. 2014, 15, 4405–4409. [Google Scholar] [CrossRef] [Green Version]
- Klochkov, S.G.; Neganova, M.E.; Yarla, N.S.; Parvathaneni, M.; Sharma, B.; Tarasov, V.V.; Barreto, G.; Bachurin, S.O.; Ashraf, G.M.; Aliev, G. Implications of farnesyltransferase and its inhibitors as a promising strategy for cancer therapy. Semin. Cancer Biol. 2019, 56, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of NrfCurr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepetsos, P.; Papavassiliou, K.A.; Papavassiliou, A.G. Redox and NF-κB signaling in osteoarthritis. Free Radic. Biol. Med. 2019, 132, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Na, H.K.; Kim, E.H.; Choi, M.A.; Park, J.M.; Kim, D.H.; Surh, Y.J. Diallyl trisulfide induces apoptosis in human breast cancer cells through ROS-mediated activation of JNK and AP-1. Biochem. Pharmacol. 2012, 84, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Klaunig, J.E.; Wang, Z.; Pu, X.; Zhou, S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol. Appl. Pharmacol. 2011, 254, 86–99. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- He, X.; Lin, G.X.; Chen, M.G.; Zhang, J.X.; Ma, Q. Protection against chromium (VI)-induced oxidative stress and apoptosis by NrfRecruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol. Sci. 2007, 98, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; De La Vega, M.R.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; De La Vega, M.R.; Chapman, E.; Ooi, A.; Zhang, D.D. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 2018, 57, 182–192. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334–351. [Google Scholar] [CrossRef] [Green Version]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 Prevents Initiation but Accelerates Progression through the Kras Signaling Pathway during Lung Carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Schiff, R.; Reddy, P.; Ahotupa, M.; Coronado-Heinsohn, E.; Grim, M.; Hilsenbeck, S.G.; Lawrence, R.; Deneke, S.; Herrera, R.; Chamness, G.C.; et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J. Natl. Cancer Inst. 2000, 92, 1926–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trop-Steinberg, S.; Azar, Y. AP-1 Expression and its Clinical Relevance in Immune Disorders and Cancer. Am. J. Med. Sci. 2017, 353, 474–483. [Google Scholar] [CrossRef]
- Bieche, I.; Lerebours, F.; Tozlu, S.; Espie, M.; Marty, M.; Lidereau, R. Molecular profiling of inflammatory breast cancer: Identification of a poor-prognosis gene expression signature. Clin. Cancer Res. 2004, 10, 6789–6795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passegué, E.; Wagner, E.F.; Weissman, I.L. JunB Deficiency Leads to a Myeloproliferative Disorder Arising from Hematopoietic Stem Cells. Cell 2004, 119, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, K.; Quintás-Cardama, A.; Radich, J.; Dai, H.; Yang, H.; Garcia-Manero, G. Downregulation of JUNB mRNA expression in advanced phase chronic myelogenous leukemia. Leuk. Res. 2009, 33, 1361–1366. [Google Scholar] [CrossRef] [Green Version]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890. [Google Scholar] [CrossRef] [Green Version]
- Rasmi, R.R.; Sakthivel, K.M.; Guruvayoorappan, C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed. Pharmacother. 2020, 130, 110569. [Google Scholar] [CrossRef]
- Ma, C.; Zu, X.; Liu, K.; Bode, A.M.; Dong, Z.; Liu, Z.; Kim, D.J. Knockdown of Pyruvate Kinase M Inhibits Cell Growth and Migration by Reducing NF-kB Activity in Triple-Negative Breast Cancer Cells. Mol. Cells 2019, 42, 628–636. [Google Scholar]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; Van Dam, P.A. The role of Nuclear Factor-κ B signaling in human cervical cancer. Crit. Rev. Oncol./Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef]
- Sokolova, O.; Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef]
- Thomas-Jardin, S.E.; Dahl, H.; Nawas, A.F.; Bautista, M.; Delk, N.A. NF-κB signaling promotes castration-resistant prostate cancer initiation and progression. Pharmacol. Ther. 2020, 211, 107538. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Di Francesco, B.; Alesse, E.; Franzoso, G.; Zazzeroni, F. NF-κB and mitochondria cross paths in cancer: Mitochondrial metabolism and beyond. Semin. Cell Dev. Biol. 2020, 98, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wu, R.; Lin, M.; Liang, Y.; Liu, J.; Wang, X.; Yang, B.; Feng, Z. RRAD inhibits the Warburg effect through negative regulation of the NF-κB signaling. Oncotarget 2015, 6, 14982–14992. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Shen, H.M.; Tergaonkar, V. NFκB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 2009, 14, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef]
- Bahrami, A.; Atkin, S.L.; Majeed, M.; Sahebkar, A. Effects of curcumin on hypoxia-inducible factor as a new therapeutic target. Pharmacol. Res. 2018, 137, 159–169. [Google Scholar] [CrossRef]
- de Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Semenza, G.L.; Zhang, H.-F. Hypoxia-inducible factor 1 and breast cancer metastasis. J. Zhejiang Univ. Sci. B 2015, 16, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Qin, Q.; Yang, M.; Zhang, H.; Liu, Z.; Yang, Y.; Chen, X.; Zhu, H.; Wang, D.; Meng, C.; et al. Bortezomib enhances the radiosensitivity of hypoxic cervical cancer cells by inhibiting HIF-1α expression. Int. J. Clin. Exp. Pathol. 2015, 8, 9032–9041. [Google Scholar] [PubMed]
- Wei, T.; Zhao, F.-Y.; Zhang, L.; Fu, Q.; Mao, M.; Mu, D.-Z.; Qu, Y. Effect of HIF-1alpha on the proliferation and apoptosis of uterine cervix cancer SiHa cells. Sichuan Da Xue Xue Bao Yi Xue Ban = J. Sichuan Univ. Med. Sci. Ed. 2008, 39, 378–382. [Google Scholar]
- Jackson, A.L.; Zhou, B.; Kim, W.Y. HIF, hypoxia and the role of angiogenesis in non-small cell lung cancer. Expert Opin. Ther. Targets 2010, 14, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Qiu, Q.; Li, Z.; Sachdeva, M.; Min, H.; Cardona, D.M.; Delaney, T.F.; Han, T.; Ma, Y.; Luo, L.; et al. HIF-1 Alpha Regulates the Response of Primary Sarcomas to Radiation Therapy through a Cell Autonomous Mechanism. Radiat. Res. 2015, 183, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Huang, H.; Liu, Q.; Zhu, L.; Zhang, Y.; Lu, X.; Wu, Y.; Liu, L. Prognostic Value of Preoperative Systemic Immune-Inflammation Index in Patients with Cervical Cancer. Sci. Rep. 2019, 9, 3284. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, G.J.; Charles, K.A.; Roxburgh, C.S.; Horgan, P.G.; McMillan, D.C.; Clarke, S.J. The systemic inflammation-based neutrophil–lymphocyte ratio: Experience in patients with cancer. Crit. Rev. Oncol. Hematol. 2013, 88, 218–230. [Google Scholar] [CrossRef]
- Rubio-Jurado, B.; Balderas-Pena, L.M.; Garcia-Luna, E.E.; Zavala-Cerna, M.G.; Riebeling-Navarro, C.; Reyes, P.A.; Nava-Zavala, A.H. Obesity, Thrombotic Risk, and Inflammation in Cancer. Adv. Clin. Chem. 2018, 85, 71–89. [Google Scholar]
- Grabosch, S.; Bulatović, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Kuprash, D.V.; Nedospasov, S.A. Molecular and cellular mechanisms of inflammation. Biochemistry 2016, 81, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Arulselvan, P.; Fard, M.T.; Tan, W.S.; Gothai, S.; Fakurazi, S.; Norhaizan, M.E.; Kumar, S.S. Role of Antioxidants and Natural Products in Inflammation. Oxidative Med. Cell. Longev. 2016, 2016, 5276130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germolec, D.R.; Shipkowski, K.A.; Frawley, R.P.; Evans, E. Markers of Inflammation. Methods Mol. Biol. 2018, 1803, 57–79. [Google Scholar] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Karin, M.; Sun, B. Targeting cancer-promoting inflammation-have anti-inflammatory therapies come of age? Nat. Rev. Clin. Oncol. 2021, 18, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Greten, F.R. Cell plasticity in epithelial homeostasis and tumorigenesis. Nature 2017, 19, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Peek, R.M., Jr. Helicobacter pylori, Cancer, and the Gastric Microbiota. Adv. Exp. Med. Biol. 2016, 908, 393–408. [Google Scholar]
- Mentis, A.-F.A.; Boziki, M.; Grigoriadis, N.; Papavassiliou, A.G. Helicobacter pylori infection and gastric cancer biology: Tempering a double-edged sword. Cell. Mol. Life Sci. 2019, 76, 2477–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugano, K. Effect of Helicobacter pylori eradication on the incidence of gastric cancer: A systematic review and meta-analysis. Gastric Cancer 2019, 22, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Salar, A. Gastric MALT lymphoma and Helicobacter pylori. Med. Clin. 2019, 152, 65–71. [Google Scholar] [CrossRef]
- Ramirez-Garcia, A.; Rementeria, A.; Aguirre-Urizar, J.M.; Moragues, M.D.; Antoran, A.; Pellon, A.; Abad-Diaz-de-Cerio, A.; Hernando, F.L. Candida albicans and cancer: Can this yeast induce cancer development or progression? Crit. Rev. Microbiol. 2016, 42, 181–193. [Google Scholar]
- Ho, J.; Camilli, G.; Griffiths, J.S.; Richardson, J.P.; Kichik, N.; Naglik, J.R. Candida albicans and candidalysin in inflammatory disorders and cancer. Immunology 2021, 162, 11–16. [Google Scholar] [CrossRef]
- Nawaz, A.; Mäkinen, A.; Pärnänen, P.; Meurman, J.H. Proteolytic activity of non-albicans Candida and Candida albicans in oral cancer patients. New Microbiol. 2018, 41, 296–301. [Google Scholar]
- Yang, S.; Zhao, W.; Wang, H.; Wang, Y.; Li, J.; Wu, X. Trichomonas vaginalis infection-associated risk of cervical cancer: A meta-analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 228, 166–173. [Google Scholar] [CrossRef]
- Tsang, S.H.; Peisch, S.F.; Rowan, B.; Markt, S.C.; Gonzalez-Feliciano, A.G.; Sutcliffe, S.; Platz, E.A.; Mucci, L.A.; Ebot, E.M. Association between Trichomonas vaginalis and prostate cancer mortality. Int. J. Cancer 2019, 144, 2377–2380. [Google Scholar] [CrossRef]
- Hemmat, N.; Bannazadeh Baghi, H. Association of human papillomavirus infection and inflammation in cervical cancer. Pathog. Dis. 2019, 77, ftz048. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Y.; Lu, X.; Zhao, H.; Chen, C.; Wang, Y.; Hu, W.; Zhu, Y.; Yan, H.; Yan, F. Genomic characterization of cervical cancer based on human papillomavirus status. Gynecol. Oncol. 2019, 152, 629–637. [Google Scholar] [CrossRef]
- Wang, R.; Pan, W.; Jin, L.; Huang, W.; Li, Y.; Wu, D.; Gao, C.; Ma, D.; Liao, S. Human papillomavirus vaccine against cervical cancer: Opportunity and challenge. Cancer Lett. 2020, 471, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cai, X.; Shen, F.; Ma, F. HPV post-infection microenvironment and cervical cancer. Cancer Lett. 2021, 497, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Stelzle, D.; Tanaka, L.F.; Lee, K.K.; Ibrahim Khalil, A.; Baussano, I.; Shah, A.S.V.; McAllister, D.A.; Gottlieb, S.L.; Klug, S.J.; Winkler, A.S.; et al. Estimates of the global burden of cervical cancer associated with HIV. Lancet Glob. Health 2021, 9, e161–e169. [Google Scholar] [CrossRef]
- Lightner, A.L.; Vogler, S.; McMichael, J.; Jia, X.; Regueiro, M.; Qazi, T.; Steele, S.R. Dysplastic Progression to Adenocarcinoma is Equivalent in Ulcerative Colitis and Crohn’s Disease. J. Crohns Colitis 2021, 15, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Dsouza, R.; Varghese, G.; Korula, D.R.; Dutta, A.K. Crohn’s disease associated adenocarcinoma of ileocaecal region: A miscalculated approach. BMJ Case Rep. 2020, 13, e234512. [Google Scholar] [CrossRef]
- Hussain, T.; Jeganathan, N.A.; Karagkounis, G.; Stocchi, L.; Shawki, S.; Holubar, S.D.; Gordon, I.; Hull, T.; Liska, D. Small bowel adenocarcinoma in Crohn’s disease: A rare but devastating complication. Tech. Coloproctol. 2020, 24, 1055–1062. [Google Scholar] [CrossRef]
- Yao, D.; Dong, M.; Dai, C.; Wu, S. Inflammation and Inflammatory Cytokine Contribute to the Initiation and Development of Ulcerative Colitis and Its Associated Cancer. Inflamm. Bowel Dis. 2019, 25, 1595–1602. [Google Scholar] [CrossRef]
- Porter, C.M.; Shrestha, E.; Peiffer, L.; Sfanos, K.S. The microbiome in prostate inflammation and prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mikrani, R.; Xie, D.; Wazir, J.; Shrestha, S.; Ullah, R.; Baig, M.M.F.A.; Ahmed, A.; Srivastava, P.K.; Thapa, K.B.; et al. Chronic prostatitis/chronic pelvic pain syndrome and prostate cancer: Study of immune cells and cytokines. Fundam. Clin. Pharmacol. 2020, 34, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2020, 56, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidzadeh, K.; Christensen, S.M.; Dalby, E.; Chandrasekaran, P.; Mosser, D.M. Macrophages and the Recovery from Acute and Chronic Inflammation. Annu. Rev. Physiol. 2017, 79, 567–592. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Cui, W.-Q.; Wei, Y.; Cui, J.; Qiu, J.; Hu, L.-L.; Gong, W.-Y.; Dong, J.-C.; Liu, B.-J. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J. Exp. Clin. Cancer Res. 2018, 37, 207. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Zhang, J.; Liang, G.; Ding, L.; He, Q.; Yang, B. Macrophage Polarization: Anti-Cancer Strategies to Target Tumor-Associated Macrophage in Breast Cancer. J. Cell. Biochem. 2017, 118, 2484–2501. [Google Scholar] [CrossRef]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef] [PubMed]
- Schwertfeger, K.L.; Xian, W.; Kaplan, A.M.; Burnett, S.H.; Cohen, D.A.; Rosen, J.M. A Critical Role for the Inflammatory Response in a Mouse Model of Preneoplastic Progression. Cancer Res. 2006, 66, 5676–5685. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Munjal, A. Interplay between inflammation and cancer. Adv. Protein Chem. Struct. Biol. 2020, 119, 199–245. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Changes in transcriptome of macrophages in atherosclerosis. J. Cell. Mol. Med. 2015, 19, 1163–1173. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Milano, E.; Pruszko, M.; Sacconi, A.; Masciarelli, S.; Iosue, I.; Melucci, E.; Gallo, E.; Terrenato, I.; Mottolese, M.; et al. Expression of ID4 protein in breast cancer cells induces reprogramming of tumour-associated macrophages. Breast Cancer Res. 2018, 20, 59. [Google Scholar] [CrossRef]
- Tan, B.; Shi, X.; Zhang, J.; Qin, J.; Zhang, N.; Ren, H.; Qian, M.; Siwko, S.; Carmon, K.; Liu, Q.; et al. Inhibition of Rspo-Lgr4 Facilitates Checkpoint Blockade Therapy by Switching Macrophage Polarization. Cancer Res. 2018, 78, 4929–4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa-Wejksza, K.; Kandefer-Szerszeń, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Han, K.; Lin, G.; Liu, C.; Wang, S.; Shi, X.; Hu, Z.; Wu, C.; Xu, X.; Hu, C. Ctenopharyngodon Idella STAT3 alleviates autophagy by up-regulating BCL-2 expression. Fish Shellfish. Immunol. 2019, 91, 194–201. [Google Scholar] [CrossRef]
- Jiao, Y.; Ding, H.; Huang, S.; Liu, Y.; Sun, X.; Wei, W.; Ma, J.; Zheng, F. Bcl-XL and Mcl-1 upregulation by calreticulin promotes apoptosis resistance of fibroblast-like synoviocytes via activation of PI3K/Akt and STAT3 pathways in rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 841–849. [Google Scholar]
- Fu, L.-Q.; Du, W.-L.; Cai, M.-H.; Yao, J.-Y.; Zhao, Y.-Y.; Mou, X.-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell. Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Granot, Z.; Fridlender, Z.G. The diversity of circulating neutrophils in cancer. Immunobiology 2017, 222, 82–88. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Paolino, G.; Corsetti, P.; Moliterni, E.; Corsetti, S.; Didona, D.; Albanesi, M.; Mattozzi, C.; Lido, P.; Calvieri, S. Mast cells and cancer. G Ital. Dermatol. Venereol. 2019, 154, 650–668. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.C.S.; Utikal, J.; Umansky, V. Opposing roles of eosinophils in cancer. Cancer Immunol. Immunother. 2019, 68, 823–833. [Google Scholar] [CrossRef]
- Cavaillon, J.-M. Exotoxins and endotoxins: Inducers of inflammatory cytokines. Toxicon 2018, 149, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, S. Pharmaceutical aspects of anti-inflammatory TNF-blocking drugs. Inflammopharmacology 2015, 23, 71–77. [Google Scholar] [CrossRef]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef]
- Ma, Y.; Ren, Y.; Dai, Z.-J.; Wu, C.-J.; Ji, Y.-H.; Xu, J. IL-6, IL-8 and TNF-α levels correlate with disease stage in breast cancer patients. Adv. Clin. Exp. Med. 2017, 26, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Nardone, B.; Orrell, K.A.; Vakharia, P.P.; West, D.P. Skin cancer associated with commonly prescribed drugs: Tumor necrosis factor alpha inhibitors (TNF-alphaIs), angiotensin-receptor blockers (ARBs), phosphodiesterase type 5 inhibitors (PDE5Is) and statins -weighing the evidence. Expert Opin. Drug Saf. 2018, 17, 139–147. [Google Scholar] [CrossRef]
- Gong, K.; Guo, G.; Gerber, D.E.; Gao, B.; Peyton, M.; Huang, C.; Minna, J.D.; Hatanpaa, K.J.; Kernstine, K.; Cai, L.; et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J. Clin. Investig. 2018, 128, 2500–2518. [Google Scholar] [CrossRef] [Green Version]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Segui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [Green Version]
- Yoshimatsu, Y.; Wakabayashi, I.; Kimuro, S.; Takahashi, N.; Takahashi, K.; Kobayashi, M.; Maishi, N.; Podyma-Inoue, K.A.; Hida, K.; Miyazono, K.; et al. TNF-alpha enhances TGF-beta-induced endothelial-to-mesenchymal transition via TGF-beta signal augmentation. Cancer Sci. 2020, 111, 2385–2399. [Google Scholar] [CrossRef]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-α is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Niu, J.; Li, M.; Teng, Y.; Wang, H.; Zhang, Y. Tumor Vasculature-Targeted Recombinant Mutated Human TNF-α Enhanced the Antitumor Activity of Doxorubicin by Increasing Tumor Vessel Permeability in Mouse Xenograft Models. PLoS ONE 2014, 9, e87036. [Google Scholar] [CrossRef] [Green Version]
- van der Veen, A.H.; de Wilt, J.H.; Eggermont, A.M.; van Tiel, S.T.; Seynhaeve, A.L.; ten Hagen, T.L. TNF- α augments intratumoural concentrations of doxorubicin in TNF- α -based isolated limb perfusion in rat sarcoma models and enhances anti-tumour effects. Br. J. Cancer 2000, 82, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Chi, W.-H.; Wang, J.; Tang, J.-J.; Lu, Y.-J. TNF-α promotes Doxorubicin-induced cell apoptosis and anti-cancer effect through downregulation of p21 in p53-deficient tumor cells. Biochem. Biophys. Res. Commun. 2005, 330, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Kuemmerle, J.F. Synergistic regulation of NOS II expression by IL-1 beta and TNF-alpha in cultured rat colonic smooth muscle cells. Am. J. Physiol. 1998, 274, G178–G185. [Google Scholar]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [Green Version]
- Lou, C.; Deng, A.; Zheng, H.; Sun, G.; Zhao, H.; Li, A.; Liu, Q.; Li, Y.; Lv, Z. Pinitol suppresses TNF-α-induced chondrocyte senescence. Cytokine 2020, 130, 155047. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-α are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef]
- Moon, D.O.; Kim, M.O.; Lee, J.D.; Choi, Y.H.; Kim, G.Y. Rosmarinic acid sensitizes cell death through suppression of TNF-alpha-induced NF-κB activation and ROS generation in human leukemia U937 cells. Cancer Lett. 2010, 288, 183–191. [Google Scholar] [CrossRef]
- Safari, H.; Zabihi, E.; Pouramir, M.; Morakabati, P.; Abedian, Z.; Karkhah, A.; Nouri, H.R. Decrease of intracellular ROS by arbutin is associated with apoptosis induction and downregulation of IL-1beta and TNF-alpha in LNCaP; prostate cancer. J. Food Biochem. 2020, 44, e13360. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Bradbury, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016, 7, 75571–75584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef]
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr.-Relat. Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Shu, B.; Yang, J.; Liu, J.; Xi, T.; Xing, Y. C-reactive protein, interleukin-6 and the risk of colorectal cancer: A meta-analysis. Cancer Causes Control. 2014, 25, 1397–1405. [Google Scholar] [CrossRef]
- Wu, D.; Wu, P.; Huang, Q.; Liu, Y.; Ye, J.; Huang, J. Interleukin-17: A promoter in colorectal cancer progression. Clin. Dev. Immunol. 2013, 2013, 436307. [Google Scholar] [CrossRef] [Green Version]
- Franzè, E.; Marafini, I.; Troncone, E.; Salvatori, S.; Monteleone, G. Interleukin-34 promotes tumorigenic signals for colon cancer cells. Cell Death Discov. 2021, 7, 245. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell. Endocrinol. 2018, 462 Pt A, 25–30. [Google Scholar] [CrossRef]
- Liu, X.; Hansen, D.M.; Timko, N.J.; Zhu, Z.; Ames, A.; Qin, C.; Nicholl, M.B.; Bai, Q.; Chen, X.; Wakefield, M.R.; et al. Association between interleukin-33 and ovarian cancer. Oncol. Rep. 2019, 41, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.M.; Gaire, B.; Vancurova, I. Interleukin-8 Induces Proliferation of Ovarian Cancer Cells in 3D Spheroids. Methods Mol. Biol. 2020, 2108, 117–124. [Google Scholar] [CrossRef]
- Pettersen, K.; Andersen, S.; van der Veen, A.; Nonstad, U.; Hatakeyama, S.; Lambert, C.; Lach-Trifilieff, E.; Moestue, S.A.; Kim, J.; Grønberg, B.H.; et al. Autocrine activin A signalling in ovarian cancer cells regulates secretion of interleukin 6, autophagy, and cachexia. J. Cachex-Sarcopenia Muscle 2020, 11, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Bai, Y.; Shen, W.; Zhao, J. Research progress on interleukin-6 in lung cancer. Zhejiang da xue xue bao. Yi Xue Ban = J. Zhejiang Univ. Med. Sci. 2018, 47, 659–664. [Google Scholar]
- Wu, F.; Xu, J.; Huang, Q.; Han, J.; Duan, L.; Fan, J.; Lv, Z.; Guo, M.; Hu, G.; Chen, L.; et al. The Role of Interleukin-17 in Lung Cancer. Mediat. Inflamm. 2016, 2016, 8494079. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1beta by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Res. 2020, 80, 5597–5605. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The role of interleukin-18 in pancreatitis and pancreatic cancer. Cytokine Growth Factor Rev. 2019, 50, 1–12. [Google Scholar] [CrossRef]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Luan, S.; An, Z.; Bi, S.; Chen, L.; Fan, J. Interleukin 6 receptor (IL-6R) was an independent prognostic factor in cervical cancer. Histol. Histopathol. 2018, 33, 269–276. [Google Scholar]
- Du, G.H.; Wang, J.K.; Richards, J.R.; Wang, J.J. Genetic polymorphisms in tumor necrosis factor alpha and interleukin-10 are associated with an increased risk of cervical cancer. Int. Immunopharmacol. 2019, 66, 154–161. [Google Scholar] [CrossRef]
- Jiang, H.; Yin, X.F.; Yu, J.Y.; Su, C.Y. The implication of interleukin-1beta in the development and progression of multiple myeloma. J. Biol. Regul. Homeost Agents 2020, 34, 547–552. [Google Scholar] [PubMed]
- Kohsari, M.; Khadem-Ansari, M.-H.; Rasmi, Y.; Sayyadi, H.; Mofrad, M.G.; Kazeminezhad, B.; Faghihloo, E. Serum Levels of Interleukin-8 and Soluble Interleukin-6 Receptor in Patients with Stage-I Multiple Myeloma: A Case-Control Study. Asian Pac. J. Cancer Prev. 2020, 21, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, I.-H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.-M.; Choi, K.S.; Son, S.-Y.; Han, S.-U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Ghandadi, M.; Sahebkar, A. Interleukin-6: A Critical Cytokine in Cancer Multidrug Resistance. Curr. Pharm. Des. 2016, 22, 518–526. [Google Scholar] [CrossRef]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef]
- Rao, Z.-L.; Cao, H.-J.; Shi, B.-Y.; Luo, J.; Liu, X.-B.; Zeng, N. Effects of Jingfang n-butanol extraction isolated fraction A on LPS-induced inflammation in RAW264.7 cells. China J. Chin. Mater. Med. 2019, 44, 1026–1033. [Google Scholar]
- Qiu, X.; Cheng, J.-C.; Chang, H.-M.; Leung, P.C.K. COX2 and PGE2 mediate EGF-induced E-cadherin-independent human ovarian cancer cell invasion. Endocr.-Relat. Cancer 2014, 21, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Raj, V.; Bhadauria, A.S.; Singh, A.K.; Kumar, U.; Rai, A.; Keshari, A.K.; Kumar, P.; Kumar, D.; Maity, B.; Nath, S.; et al. Novel 1,3,4-thiadiazoles inhibit colorectal cancer via blockade of IL-6/COX-2 mediated JAK2/STAT3 signals as evidenced through data-based mathematical modeling. Cytokine 2019, 118, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, A.J.; Lippman, S.M.; Mann, J.R.; Subbaramaiah, K.; Dubois, R.N. Cyclooxygenase-2 and Epidermal Growth Factor Receptor: Pharmacologic Targets for Chemoprevention. J. Clin. Oncol. 2005, 23, 254–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortezaee, K. Human hepatocellular carcinoma: Protection by melatonin. J. Cell. Physiol. 2018, 233, 6486–6508. [Google Scholar] [CrossRef]
- Lacy, P. Editorial: Secretion of Cytokines and Chemokines by Innate Immune Cells. Front. Immunol. 2015, 6, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.-L.; Wang, C.-S.; Huang, Y.-H.; Tsai, M.-M.; Liang, Y.; Lin, K.-H. Overexpression of CXCL1 and its receptor CXCR2 promote tumor invasion in gastric cancer. Ann. Oncol. 2011, 22, 2267–2276. [Google Scholar] [CrossRef]
- Singh, S.; Singh, A.P.; Sharma, B.; Owen, L.B.; Singh, R.K. CXCL8 and its cognate receptors in melanoma progression and metastasis. Futur. Oncol. 2010, 6, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Chen, R.; Jin, R.; Huang, Z. The role of CXCL chemokine family in the development and progression of gastric cancer. Int. J. Clin. Exp. Pathol. 2020, 13, 484–492. [Google Scholar]
- Pączek, S.; Łukaszewicz-Zając, M.; Gryko, M.; Mroczko, P.; Kulczynska-Przybik, A.; Mroczko, B. CXCL-8 in Preoperative Colorectal Cancer Patients: Significance for Diagnosis and Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2040. [Google Scholar] [CrossRef] [Green Version]
- Łukaszewicz-Zając, M.; Pączek, S.; Muszyński, P.; Kozłowski, M.; Mroczko, B. Comparison between clinical significance of serum CXCL-8 and classical tumor markers in oesophageal cancer (OC) patients. Clin. Exp. Med. 2019, 19, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Singh, S.; Varney, M.L.; Kindle, S.; Singh, R.K. Modulation of CXCL-8 expression in human melanoma cells regulates tumor growth, angiogenesis, invasion, and metastasis. Cancer Med. 2012, 1, 306–317. [Google Scholar] [CrossRef]
- Zhang, G.; Luo, X.; Zhang, W.; Chen, E.; Xu, J.; Wang, F.; Cao, G.; Ju, Z.; Jin, D.; Huang, X.; et al. CXCL-13 Regulates Resistance to 5-Fluorouracil in Colorectal Cancer. Cancer Res. Treat. 2020, 52, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xu, Z.Q.; Zong, Y.P.; Ou, B.C.; Shen, X.H.; Feng, H.; Zheng, M.H.; Zhao, J.K.; Lu, A.G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019, 10, 178. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Fan, H.; Lv, X.; Chen, S.; Shao, Z. Prognostic significance of CXCL5 expression in cancer patients: A meta-analysis. Cancer Cell Int. 2018, 18, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visekruna, A.; Volkov, A.; Steinhoff, U. A key role for NF-κB transcription factor c-Rel in T-lymphocyte-differentiation and effector functions. Clin. Dev. Immunol. 2012, 2012, 239368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, R.; Ma, L.; Zheng, Y. Anti-inflammatory effects of luteolin on acute gouty arthritis rats via TLR/MyD88/NF-κB pathway. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2020, 45, 115–122. [Google Scholar] [PubMed]
- Wu, S.; Li, H.; Yu, L.; Wang, N.; Li, X.; Chen, W. IL-1beta upregulates Muc5ac expression via NF-κB-induced HIF-1alpha in asthma. Immunol. Lett. 2017, 192, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtois, G.; Gilmore, T.D. Mutations in the NF-κB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [Green Version]
- Timucin, A.C.; Basaga, H. Pro-apoptotic effects of lipid oxidation products: HNE at the crossroads of NF-κB pathway and anti-apoptotic Bcl-Free Radic. Biol. Med. 2017, 111, 209–218. [Google Scholar] [CrossRef]
- Van Der Heijden, M.; Zimberlin, C.D.; Nicholson, A.M.; Colak, S.; Kemp, R.; Meijer, S.L.; Medema, J.P.; Greten, F.R.; Jansen, M.; Winton, D.J.; et al. Bcl-2 is a critical mediator of intestinal transformation. Nat. Commun. 2016, 7, 10916. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.J.; Mijatov, B.; Gunatilake, D.; Gowrishankar, K.; Tiffen, J.; James, W.; Jin, L.; Pupo, G.; Cullinane, C.; McArthur, G.A.; et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET Pigment. Cell Melanoma Res. 2014, 27, 1126–1137. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-κB and STAT3-key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Siersbaek, R.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.P.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor alpha Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423 e9. [Google Scholar] [CrossRef]
- Wang, X.; Shao, X.; Gu, L.; Jiang, K.; Wang, S.; Chen, J.; Fang, J.; Guo, X.; Yuan, M.; Shi, J.; et al. Targeting STAT3 enhances NDV-induced immunogenic cell death in prostate cancer cells. J. Cell. Mol. Med. 2020, 24, 4286–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yu, W.; Lian, J.; Wu, Q.; Liu, S.; Yang, L.; Li, F.; Huang, L.; Chen, X.; Zhang, Z.; et al. Th17 cells inhibit CD8+ T cell migration by systematically downregulating CXCR3 expression via IL-17A/STAT3 in advanced-stage colorectal cancer patients. J. Hematol. Oncol. 2020, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Zhao, G.; Zhang, X.; Hao, M.; Hassan, S.; Zhang, M.; Zheng, H.; Yang, D.; Liu, L.; et al. IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma. Cancer Lett. 2020, 477, 19–30. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Nie, J.; Huang, J.-P.; Zheng, G.-J.; Feng, B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomed. Pharmacother. 2019, 117, 109135. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zheng, X.; Fu, B.; Nian, Z.; Qian, Y.; Sun, R.; Tian, Z.; Wei, H. Hepatectomy promotes recurrence of liver cancer by enhancing IL-11-STAT3 signaling. EBioMedicine 2019, 46, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Kerber, E.L.; Padberg, C.; Koll, N.; Schuetzhold, V.; Fandrey, J.; Winning, S. The Importance of Hypoxia-Inducible Factors (HIF-1 and HIF-2) for the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 8551. [Google Scholar] [CrossRef] [PubMed]
- Befani, C.; Liakos, P. The role of hypoxia-inducible factor-2 alpha in angiogenesis. J. Cell Physiol. 2018, 233, 9087–9098. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage Expression of Hypoxia-Inducible Factor-1α Suppresses T-Cell Function and Promotes Tumor Progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.; Murdoch, C. Macrophage Responses to Hypoxia: Implications for Tumor Progression and Anti-Cancer Therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Wong, C.C.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.M.; Khoo, U.S.; Ng, I.O.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [Green Version]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Marelli, G.; Sica, A.; Vannucci, L.; Allavena, P. Inflammation as target in cancer therapy. Curr. Opin. Pharmacol. 2017, 35, 57–65. [Google Scholar] [CrossRef]
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007, 42, 153–164. [Google Scholar] [CrossRef]
- Dechakhamphu, S.; Pinlaor, S.; Sitthithaworn, P.; Nair, J.; Bartsch, H.; Yongvanit, P. Lipid Peroxidation and Etheno DNA Adducts in White Blood Cells of Liver Fluke-Infected Patients: Protection by Plasma α-Tocopherol and Praziquantel. Cancer Epidemiol. Biomark. Prev. 2010, 19, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Zheng, S.; Liu, H.; Li, S. Protective effects of taurine against inflammation, apoptosis, and oxidative stress in brain injury. Mol. Med. Rep. 2018, 18, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 413. [Google Scholar]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Song, Y.S. Stress Response, Inflammaging, and Cancer. Inflamm. Adv. Age Nutr. Res. Clin. Interv. 2014, 5, 49–53. [Google Scholar] [CrossRef]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Civenni, G.; Shinde, D.; Zoma, M.; Albino, D.; Costales, P.; Moris, F.; Carbone, G.; Catapano, C. The multi-kinase inhibitor EC-70124 delivers a double-hit to prostate cancer stem cells interfering with both STAT3 and NF-kB signaling. Eur. Urol. Suppl. 2017, 36, e1294. [Google Scholar] [CrossRef]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, L.; Li, Y.-C.; Wu, L.; Yu, G.-T.; Zhang, W.-F.; Sun, Z.-J. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 116. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, S.-W.; Shin, Y.-H.; Lee, J.-H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Muneer, K.M.; Tamimi, I.A.; Chang, M.E.; Ata, M.O.; Yusuf, N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol. Appl. Pharmacol. 2013, 270, 70–76. [Google Scholar] [CrossRef]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.-D. IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Athreya, K.; Xavier, M.F. Antioxidants in the Treatment of Cancer. Nutr. Cancer 2017, 69, 1099–1104. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Sivinski, J.; Zhang, D.D.; Chapman, E. Targeting NRF2 to treat cancer. Semin. Cancer Biol. 2021, 76, 61–73. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709, Erratum in 2021, 20, 652. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef]
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant polyphenols in cancer treatment: Friend, foe or foil? Semin. Cancer Biol. 2017, 46, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Clifford, T.; Acton, J.P.; Cocksedge, S.P.; Davies, K.A.B.; Bailey, S.J. The effect of dietary phytochemicals on nuclear factor erythroid 2-related factor 2 (Nrf2) activation: A systematic review of human intervention trials. Mol. Biol. Rep. 2021, 48, 1745–1761. [Google Scholar] [CrossRef]
- Frank, J.; Fukagawa, N.K.; Bilia, A.R.; Johnson, E.J.; Kwon, O.; Prakash, V.; Miyazawa, T.; Clifford, M.N.; Kay, C.D.; Crozier, A.; et al. Terms and nomenclature used for plant-derived components in nutrition and related research: Efforts toward harmonization. Nutr. Rev. 2020, 78, 451–458. [Google Scholar] [CrossRef]
- Crozier, A.; Yokota, T.; Jaganath, I.B.; Marks, S.; Saltmarsh, M.; Clifford, M.N. Secondary Metabolites in Fruits, Vegetables, Beverages and Other Plant-based Dietary Components. In Plant Secondary Metabolites: Occurrence, Structure and Role in the Human Diet; Crozier, A., Clifford, M.N., Ashihara, H., Eds.; Blackwell Pub Oxford: Ames, IA, USA, 2006; pp. 208–302. [Google Scholar] [CrossRef]
- Li, G.; Ding, K.; Qiao, Y.; Zhang, L.; Zheng, L.; Pan, T.; Zhang, L. Flavonoids Regulate Inflammation and Oxidative Stress in Cancer. Molecules 2020, 25, 5628. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodor, E.D.; Ungureanu, O.; Gatea, F.; Radu, G.L. The Potential of Flavonoids and Tannins from Medicinal Plants as Anticancer Agents. Anti-Cancer Agents Med. Chem. 2020, 20, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Bisol, A.; De Campos, P.S.; Lamers, M.L. Flavonoids as anticancer therapies: A systematic review of clinical trials. Phytother. Res. 2020, 34, 568–582. [Google Scholar] [CrossRef]
- Jayasuriya, R.; Dhamodharan, U.; Ali, D.; Ganesan, K.; Xu, B.; Ramkumar, K.M. Targeting Nrf2/Keap1 signaling pathway by bioactive natural agents: Possible therapeutic strategy to combat liver disease. Phytomedicine 2021, 92, 153755. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Activation of Nrf2 by Natural Bioactive Compounds: A Promising Approach for Stroke? Int. J. Mol. Sci. 2020, 21, 4875. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent Advances of Natural Polyphenols Activators for Keap1-Nrf2 Signaling Pathway. Chem. Biodivers. 2019, 16, e1900400. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [Green Version]
- El-Seedi, H.R.; Yosri, N.; Khalifa, S.A.M.; Guo, Z.; Musharraf, S.G.; Xiao, J.; Saeed, A.; Du, M.; Khatib, A.; Abdel-Daim, M.M.; et al. Exploring natural products-based cancer therapeutics derived from egyptian flora. J. Ethnopharmacol. 2021, 269, 113626. [Google Scholar] [CrossRef]
- Ornano, L.; Venditti, A.; Donno, Y.; Sanna, C.; Ballero, M.; Bianco, A. Phytochemical analysis of non-volatile fraction of Artemisia caerulescens subsp. densiflora (Viv.) (Asteraceae), an endemic species of La Maddalena Archipelago (Sardinia–Italy). Nat. Prod. Res. 2016, 30, 920–925. [Google Scholar] [CrossRef]
- Ayoobi, F.; Shamsizadeh, A.; Fatemi, I.; Vakilian, A.; Allahtavakoli, M.; Hassanshahi, G.; Moghadam-Ahmadi, A. Bio-effectiveness of the main flavonoids of Achillea millefolium in the pathophysiology of neurodegenerative disorders—A review. Iran J. Basic Med. Sci. 2017, 20, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Yapıcı, I.; Altay, A.; Ozturk Sariıkaya, B.; Korkmaz, M.; Atila, A.; Gülçin, I.; Köksal, E. In vitro Antioxidant and Cytotoxic Activities of Extracts of Endemic Tanacetum erzincanense Together with Phenolic Content by LC-ESI-QTOF-MS. Chem. Biodivers. 2021, 18, e2000812. [Google Scholar] [CrossRef] [PubMed]
- Ginwala, R.; Bhavsar, R.; Chigbu, D.G.I.; Jain, P.; Khan, Z.K. Potential Role of Flavonoids in Treating Chronic Inflammatory Diseases with a Special Focus on the Anti-Inflammatory Activity of Apigenin. Antioxidants 2019, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, P.; Shikha, D.; Thakur, M.; Aneja, A. Functionality of apigenin as a potent antioxidant with emphasis on bioavailability, metabolism, action mechanism and in vitro and in vivo studies: A review. J. Food Biochem. 2021, e13950. [Google Scholar] [CrossRef] [PubMed]
- Maggioni, D.; Garavello, W.; Rigolio, R.; Pignataro, L.; Gaini, R.; Nicolini, G. Apigenin impairs oral squamous cell carcinoma growth in vitro inducing cell cycle arrest and apoptosis. Int. J. Oncol. 2013, 43, 1675–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizumi, Y.; Oishi, M.; Taniguchi, T.; Goi, W.; Sowa, Y.; Sakai, T. The Flavonoid Apigenin Downregulates CDK1 by Directly Targeting Ribosomal Protein S9. PLoS ONE 2013, 8, e73219. [Google Scholar] [CrossRef]
- Seo, H.S.; Choi, H.S.; Kim, S.R.; Choi, Y.K.; Woo, S.M.; Shin, I.; Woo, J.K.; Park, S.Y.; Shin, Y.C.; Ko, S.G. Apigenin induces apoptosis via extrinsic pathway, inducing p53 and inhibiting STAT3 and NFκB signaling in HER2-overexpressing breast cancer cells. Mol. Cell Biochem. 2012, 366, 319–334. [Google Scholar] [CrossRef]
- Imran, M.; Aslam Gondal, T.; Atif, M.; Shahbaz, M.; Batool Qaisarani, T.; Hanif Mughal, M.; Salehi, B.; Martorell, M.; Sharifi-Rad, J. Apigenin as an anticancer agent. Phytother. Res. 2020, 34, 1812–1828. [Google Scholar] [CrossRef]
- Liu, J.; Cao, X.-C.; Xiao, Q.; Quan, M.-F. Apigenin inhibits HeLa sphere-forming cells through inactivation of casein kinase 2α. Mol. Med. Rep. 2015, 11, 665–669. [Google Scholar] [CrossRef]
- Silva-Pavez, E.; Tapia, J.C. Protein Kinase CK2 in Cancer Energetics. Front. Oncol. 2020, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.-W.; Chiang, L.-C.; Lin, C.-C. Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci. 2005, 76, 1367–1379. [Google Scholar] [CrossRef]
- Zhang, Y.; Tighe, S.; Zhu, Y.-T. COX-2 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 87–104. [Google Scholar] [CrossRef]
- Lau, G.T.Y.; Leung, L.K. The dietary flavonoid apigenin blocks phorbol 12-myristate 13-acetate-induced COX-2 transcriptional activity in breast cell lines. Food Chem. Toxicol. 2010, 48, 3022–3027. [Google Scholar] [CrossRef]
- Woo, J.S.; Choo, G.S.; Yoo, E.S.; Kim, S.H.; Lee, J.H.; Han, S.H.; Kim, H.J.; Jung, S.H.; Park, Y.S.; Kim, B.S.; et al. Apigenin induces apoptosis by regulating Akt and MAPK pathways in human melanoma cell A375SM. Mol. Med. Rep. 2020, 22, 4877–4889. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, M.; Liu, Y.; Zhang, Z.; Lu, R.; Lu, J. Apigenin inhibits cell proliferation, migration, and invasion by targeting Akt in the A549 human lung cancer cell line. Anti-Cancer Drugs 2017, 28, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Adham, A.N.; Abdelfatah, S.; Naqishbandi, A.M.; Mahmoud, N.; Efferth, T. Cytotoxicity of apigenin toward multiple myeloma cell lines and suppression of iNOS and COX-2 expression in STAT1-transfected HEK293 cells. Phytomedicine 2021, 80, 153371. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.Y.; Qin, Y.; Liu, H.J.; Cui, Z.H.; Li, M.; Yang, J.H.; Zhong, W.L.; Liu, Y.R.; Chen, S.; Sun, T.; et al. Apigenin inhibits colonic inflammation and tumorigenesis by suppressing STAT3-NF-κB signaling. Oncotarget 2017, 8, 100216–100226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, D.; Mazzio, E.; Soliman, K.F.A. Whole Transcriptomic Analysis of Apigenin on TNFalpha Immuno-activated MDA-MB-231 Breast Cancer Cells. Cancer Genom. Proteom. 2019, 16, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Dashwood, R.H. Epigenetic Regulation of NRF2/KEAP1 by Phytochemicals. Antioxidants 2020, 9, 865. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gonzalez, X.; Fuentes, F.; Su, Z.-Y.; Kong, A.-N.T. Apigenin Reactivates Nrf2 Anti-oxidative Stress Signaling in Mouse Skin Epidermal JB6 P + Cells Through Epigenetics Modifications. AAPS J. 2014, 16, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, H.; Wu, X.; Fang, J. Therapeutic Effects of Quercetin on Inflammation, Obesity, and Type 2 Diabetes. Mediat. Inflamm. 2016, 2016, 9340637. [Google Scholar] [CrossRef]
- Williamson, G.; Manach, C. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am. J. Clin. Nutr. 2005, 81 (Suppl. S1), 243S–255S. [Google Scholar] [CrossRef]
- Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef] [Green Version]
- Maleki Dana, P.; Sadoughi, F.; Asemi, Z.; Yousefi, B. Anti-cancer properties of quercetin in osteosarcoma. Cancer Cell Int. 2021, 21, 349. [Google Scholar] [CrossRef]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxidative Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marunaka, Y.; Marunaka, R.; Sun, H.; Yamamoto, T.; Kanamura, N.; Inui, T.; Taruno, A. Actions of Quercetin, a Polyphenol, on Blood Pressure. Molecules 2017, 22, 209. [Google Scholar] [CrossRef]
- Manjeet, K.R.; Ghosh, B. Quercetin inhibits LPS-induced nitric oxide and tumor necrosis factor-alpha production in murine macrophages. Int. J. Immunopharmacol. 1999, 21, 435–443. [Google Scholar]
- Bureau, G.; Longpré, F.; Martinoli, M.-G. Resveratrol and quercetin, two natural polyphenols, reduce apoptotic neuronal cell death induced by neuroinflammation. J. Neurosci. Res. 2008, 86, 403–410. [Google Scholar] [CrossRef]
- Kim, H.P.; Mani, I.; Iversen, L.; Ziboh, V.A. Effects of naturally-occurring flavonoids and biflavonoids on epidermal cyclooxygenase and lipoxygenase from guinea-pigs. Prostaglandins Leukot. Essent. Fat. Acids 1998, 58, 17–24. [Google Scholar] [CrossRef]
- Han, M.; Song, Y.; Zhang, X. Quercetin Suppresses the Migration and Invasion in Human Colon Cancer Caco-2 Cells Through Regulating Toll-like Receptor 4/Nuclear Factor-κ B Pathway. Pharmacogn. Mag. 2016, 12 (Suppl. S2), S237–S244. [Google Scholar]
- Ranganathan, S.; Halagowder, D.; Sivasithambaram, N.D. Quercetin Suppresses Twist to Induce Apoptosis in MCF-7 Breast Cancer Cells. PLoS ONE 2015, 10, e0141370. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-S.; Ku, J.M.; Choi, H.-S.; Choi, Y.K.; Woo, J.-K.; Kim, M.; Kim, I.; Na, C.H.; Hur, H.; Jang, B.-H.; et al. Quercetin induces caspase-dependent extrinsic apoptosis through inhibition of signal transducer and activator of transcription 3 signaling in HER2-overexpressing BT-474 breast cancer cells. Oncol. Rep. 2016, 36, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Levesque, J.; Bousquet-Gagnon, N.; Beliveau, R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp. Cell Res. 2012, 318, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Lee, S.J.; Choi, Y.J.; Kim, M.J.; Kim, T.Y.; Ko, S.G. Quercetin Induces Apoptosis in Glioblastoma Cells by Suppressing Axl/IL-6/STAT3 Signaling Pathway. Am. J. Chin. Med. 2021, 49, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Si, Y.; Wang, Z.; Wang, J.; Guo, Y.; Zhang, X. Quercetin inhibits the growth of human gastric cancer stem cells by inducing mitochondrial-dependent apoptosis through the inhibition of PI3K/Akt signaling. Int. J. Mol. Med. 2016, 38, 619–626. [Google Scholar] [CrossRef]
- Ward, A.B.; Mir, H.; Kapur, N.; Gales, D.N.; Carriere, P.P.; Singh, S. Quercetin inhibits prostate cancer by attenuating cell survival and inhibiting anti-apoptotic pathways. World J. Surg. Oncol. 2018, 16, 108. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Huang, W.C.; JH, S.P.; Wu, Y.H.; Cheng, C.Y. Quercetin Inhibits the Production of IL-1beta-Induced Inflammatory Cytokines and Chemokines in ARPE-19 Cells via the MAPK and NF-κB Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 2957. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.C.; Chan, S.T.; Chang, C.N.; Yu, P.S.; Chuang, C.H.; Yeh, S.L. Quercetin and chrysin inhibit nickel-induced invasion and migration by downregulation of TLR4/NF-κB signaling in A549cells. Chem. Biol. Interact 2018, 292, 101–109. [Google Scholar] [CrossRef]
- Sharma, A.; Parikh, M.; Shah, H.; Gandhi, T. Modulation of Nrf2 by quercetin in doxorubicin-treated rats. Heliyon 2020, 6, e03803. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Tan, L.; Wang, M.; Ren, C.; Guo, C.; Yang, B.; Ren, Y.; Cao, Z.; Li, Y.; Pei, J. Myricetin: A review of the most recent research. Biomed. Pharmacother. 2021, 134, 111017. [Google Scholar] [CrossRef]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients 2016, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, X.; Liu, B.; Guo, W.; Wei, L.; Lin, Y.; Guo, Y.; Gong, Q.; Li, Y.; Xu, D.; Cao, Y.; et al. Myricetin relieves LPS-induced mastitis by inhibiting inflammatory response and repairing the blood-milk barrier. J. Cell. Physiol. 2019, 234, 16252–16262. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Huang, D.; Sun, C.; Xie, J.; Chen, T.; Zhao, X.; Huang, Y.; Li, D.; Wu, B.; et al. Myricetin inhibits TNF-alpha-induced inflammation in A549 cells via the SIRT1/NF-κB pathway. Pulm. Pharmacol. Ther. 2020, 65, 102000. [Google Scholar] [CrossRef]
- da Lee, H.; Lee, C.S. Flavonoid myricetin inhibits TNF-alpha-stimulated production of inflammatory mediators by suppressing the Akt, mTOR and NF-κB pathways in human keratinocytes. Eur. J. Pharmacol. 2016, 784, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Feng, Z.; Li, Q.; Qi, Z.; Zhang, Y. Myricitrin Modulates NADPH Oxidase-Dependent ROS Production to Inhibit Endotoxin-Mediated Inflammation by Blocking the JAK/STAT1 and NOX2/p47(phox) Pathways. Oxidative Med. Cell Longev. 2017, 2017, 9738745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, S.M.; Khalaf, M.M.; Sadek, S.A.; Abo-Youssef, A.M. Protective effects of apigenin and myricetin against cisplatin-induced nephrotoxicity in mice. Pharm. Biol. 2017, 55, 766–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.O.; Yin, H.H.; Park, S.H.; Byun, E.B.; Ha, H.Y.; Jang, S.I. Anti-inflammatory activity of myricetin from Diospyros lotus through suppression of NF-κB and STAT1 activation and Nrf2-mediated HO-1 induction in lipopolysaccharide-stimulated RAW264.7 macrophages. Biosci. Biotechnol. Biochem. 2016, 80, 1520–1530. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cui, S.-X.; Sun, S.-Y.; Shi, W.-N.; Song, Z.-Y.; Wang, S.-Q.; Yu, X.-F.; Gao, Z.-H.; Qu, X.-J. Chemoprevention of intestinal tumorigenesis by the natural dietary flavonoid myricetin in APCMin/+ mice. Oncotarget 2016, 7, 60446–60460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.-L.; Zhang, P.-M.; Jiang, M.; Yu, S.-W.; Wang, L. Myricetin induces apoptosis and autophagy by inhibiting PI3K/Akt/mTOR signalling in human colon cancer cells. BMC Complement. Med. Ther. 2020, 20, 209. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Yun, S.M.; Song, M.Y.; Jung, K.; Kim, E.H. Cyanidin Chloride Induces Apoptosis by Inhibiting NF-κB Signaling through Activation of Nrf2 in Colorectal Cancer Cells. Antioxidants 2020, 9, 285. [Google Scholar] [CrossRef] [Green Version]
- Straßmann, S.; Brehmer, T.; Passon, M.; Schieber, A. Methylation of Cyanidin-3-O-Glucoside with Dimethyl Carbonate. Molecules 2021, 26, 1342. [Google Scholar] [CrossRef]
- Rahman, S.; Mathew, S.; Nair, P.; Ramadan, W.S.; Vazhappilly, C.G. Health benefits of cyanidin-3-glucoside as a potent modulator of Nrf2-mediated oxidative stress. Inflammopharmacology 2021, 29, 907–923. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, S.; Zhao, X.; Ni, H.; Song, X.; Wang, W.; Yao, L.; Zhao, X.; Fu, Y. Cyanidin-3-glucoside protects liver from oxidative damage through AMPK/Nrf2 mediated signaling pathway in vivo and in vitro. J. Funct. Foods 2020, 73, 104148. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Pezzani, R.; Redaelli, M.; Zorzan, M.; Imran, M.; Ahmed Khalil, A.; Salehi, B.; Sharopov, F.; Cho, W.C.; Sharifi-Rad, J. Preclinical Pharmacological Activities of Epigallocatechin-3-gallate in Signaling Pathways: An Update on Cancer. Molecules 2020, 25, 467. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Taylor, L.S.; Ferruzzi, M.G.; Mauer, L.J. Kinetic Study of Catechin Stability: Effects of pH, Concentration, and Temperature. J. Agric. Food Chem. 2012, 60, 12531–12539. [Google Scholar] [CrossRef]
- Subramanian, P. Lipid-Based Nanocarrier System for the Effective Delivery of Nutraceuticals. Molecules 2021, 26, 5510. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Sun, Z.; Chen, G.; Zhang, H.; Ma, X.; Su, W.; Cui, X.; Li, X. Size-controlled, colloidally stable and functional nanoparticles based on the molecular assembly of green tea polyphenols and keratins for cancer therapy. J. Mater. Chem. B 2018, 6, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Chen, L.; Yokoyama, W.; Williams, P.A.; Zhong, F. Niosomes Consisting of Tween-60 and Cholesterol Improve the Chemical Stability and Antioxidant Activity of (−)-Epigallocatechin Gallate under Intestinal Tract Conditions. J. Agric. Food Chem. 2016, 64, 9180–9188. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Liu, Z.; Li, J.; Zhang, Q.; Zhong, P.; Teng, T.; Chen, M.; Xie, Z.; Ji, A.; Li, Y. Epigallocatechin-3-gallate inhibits the growth and increases the apoptosis of human thyroid carcinoma cells through suppression of EGFR/RAS/RAF/MEK/ERK signaling pathway. Cancer Cell Int. 2019, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ahmad, N.; Nieminen, A.-L.; Mukhtar, H. Growth Inhibition, Cell-Cycle Dysregulation, and Induction of Apoptosis by Green Tea Constituent (-)-Epigallocatechin-3-gallate in Androgen-Sensitive and Androgen-Insensitive Human Prostate Carcinoma Cells. Toxicol. Appl. Pharmacol. 2000, 164, 82–90. [Google Scholar] [CrossRef]
- Lim, Y.C.; Cha, Y.Y. Epigallocatechin-3-gallate induces growth inhibition and apoptosis of human anaplastic thyroid carcinoma cells through suppression of EGFR/ERK pathway and cyclin B1/CDK1 complex. J. Surg. Oncol. 2011, 104, 776–780. [Google Scholar] [CrossRef]
- Khan, N.; Mukhtar, H. Tea Polyphenols in Promotion of Human Health. Nutrients 2018, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018, 10, 1936. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-J.; Wang, K.-L.; Chen, H.-Y.; Chiang, Y.-F.; Hsia, S.-M. Protective Effects of Epigallocatechin Gallate (EGCG) on Endometrial, Breast, and Ovarian Cancers. Biomolecules 2020, 10, 1481. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Talebi, M.; Talebi, M.; Farkhondeh, T.; Mishra, G.; Ilgün, S.; Samarghandian, S. New insights into the role of the Nrf2 signaling pathway in green tea catechin applications. Phytother. Res. 2021, 35, 3078–3112. [Google Scholar] [CrossRef]
- Sun, W.; Liu, X.; Zhang, H.; Song, Y.; Li, T.; Liu, X.; Liu, Y.; Guo, L.; Wang, F.; Yang, T.; et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic. Biol. Med. 2017, 108, 840–857. [Google Scholar] [CrossRef]
- Snopek, L.; Mlcek, J.; Sochorova, L.; Baron, M.; Hlavacova, I.; Jurikova, T.; Kizek, R.; Sedlackova, E.; Sochor, J. Contribution of Red Wine Consumption to Human Health Protection. Molecules 2018, 23, 1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Lastra, C.A.; Villegas, I. Resveratrol as an antioxidant and pro-oxidant agent: Mechanisms and clinical implications. Biochem. Soc. Trans. 2007, 35 Pt 5, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Tejada, S.; Capó, X.; Mascaró, C.M.; Monserrat-Mesquida, M.; Quetglas-Llabrés, M.M.; Pons, A.; Tur, J.A.; Sureda, A. Hepatoprotective Effects of Resveratrol in Non-Alcoholic Fatty Live Disease. Curr. Pharm. Des. 2021, 27, 2558–2570. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [Green Version]
- Jeyaraman, M.M.; Al-Yousif, N.S.H.; Mann, A.S.; Dolinsky, V.W.; Rabbani, R.; Zarychanski, R.; Abou-Setta, A.M. Resveratrol for adults with type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2020, 1, CD011919. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Mohammadinejad, R.; Farkhondeh, T.; Samarghandian, S. Protective Effect of Resveratrol against Glioblastoma: A Review. Anti-Cancer Agents Med. Chem. 2021, 21, 1216–1227. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1428–1447. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Meng, T.; Xiao, D.; Muhammed, A.; Deng, J.; Chen, L.; He, J. Anti-Inflammatory Action and Mechanisms of Resveratrol. Molecules 2021, 26, 229. [Google Scholar] [CrossRef]
- Kabel, A.M.; Atef, A.; Estfanous, R.S. Ameliorative potential of sitagliptin and/or resveratrol on experimentally-induced clear cell renal cell carcinoma. Biomed. Pharmacother. 2018, 97, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Feng, Z.; Bergan, R.; Li, B.; Qin, Y.; Zhao, L.; Zhang, Z.; Shi, M. Involvement of the PI3K/Akt/Nrf2 Signaling Pathway in Resveratrol-Mediated Reversal of Drug Resistance in HL-60/ADR Cells. Nutr. Cancer 2019, 71, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yan, B.; Chen, K.; Jiang, Z.; Zhou, C.; Cao, J.; Qian, W.; Li, J.; Sun, L.; Ma, J.; et al. Resveratrol-Induced Downregulation of NAF-1 Enhances the Sensitivity of Pancreatic Cancer Cells to Gemcitabine via the ROS/Nrf2 Signaling Pathways. Oxidative Med. Cell. Longev. 2018, 2018, 9482018. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Bordoloi, D.; Padmavathi, G.; Monisha, J.; Roy, N.K.; Prasad, S.; Aggarwal, B.B. Curcumin, the golden nutraceutical: Multitargeting for multiple chronic diseases. Br. J. Pharmacol. 2017, 174, 1325–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Uddin, S.J.; Hasan, F.; Afroz, M.; Sarker, D.K.; Rouf, R.; Islam, M.T.; Shilpi, J.A.; Mubarak, M.S. Curcumin and its Multi-target Function Against Pain and Inflammation: An Update of Pre-clinical Data. Curr. Drug Targets 2021, 22, 656–671. [Google Scholar] [CrossRef]
- Tuorkey, M. Curcumin a potent cancer preventive agent: Mechanisms of cancer cell killing. Interv. Med. Appl. Sci. 2014, 6, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irshad, R.; Husain, M. Natural products in the reprogramming of cancer epigenetics. Toxicol. Appl. Pharmacol. 2021, 417, 115467. [Google Scholar] [CrossRef] [PubMed]
- Fabianowska-Majewska, K.; Kaufman-Szymczyk, A.; Szymanska-Kolba, A.; Jakubik, J.; Majewski, G.; Lubecka, K. Curcumin from Turmeric Rhizome: A Potential Modulator of DNA Methylation Machinery in Breast Cancer Inhibition. Nutrients 2021, 13, 332. [Google Scholar] [CrossRef]
- Hassan, F.U.; Rehman, M.S.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Gupta, S.C.; Sung, B. Curcumin: An orally bioavailable blocker of TNF and other pro-inflammatory biomarkers. Br. J. Pharmacol. 2013, 169, 1672–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
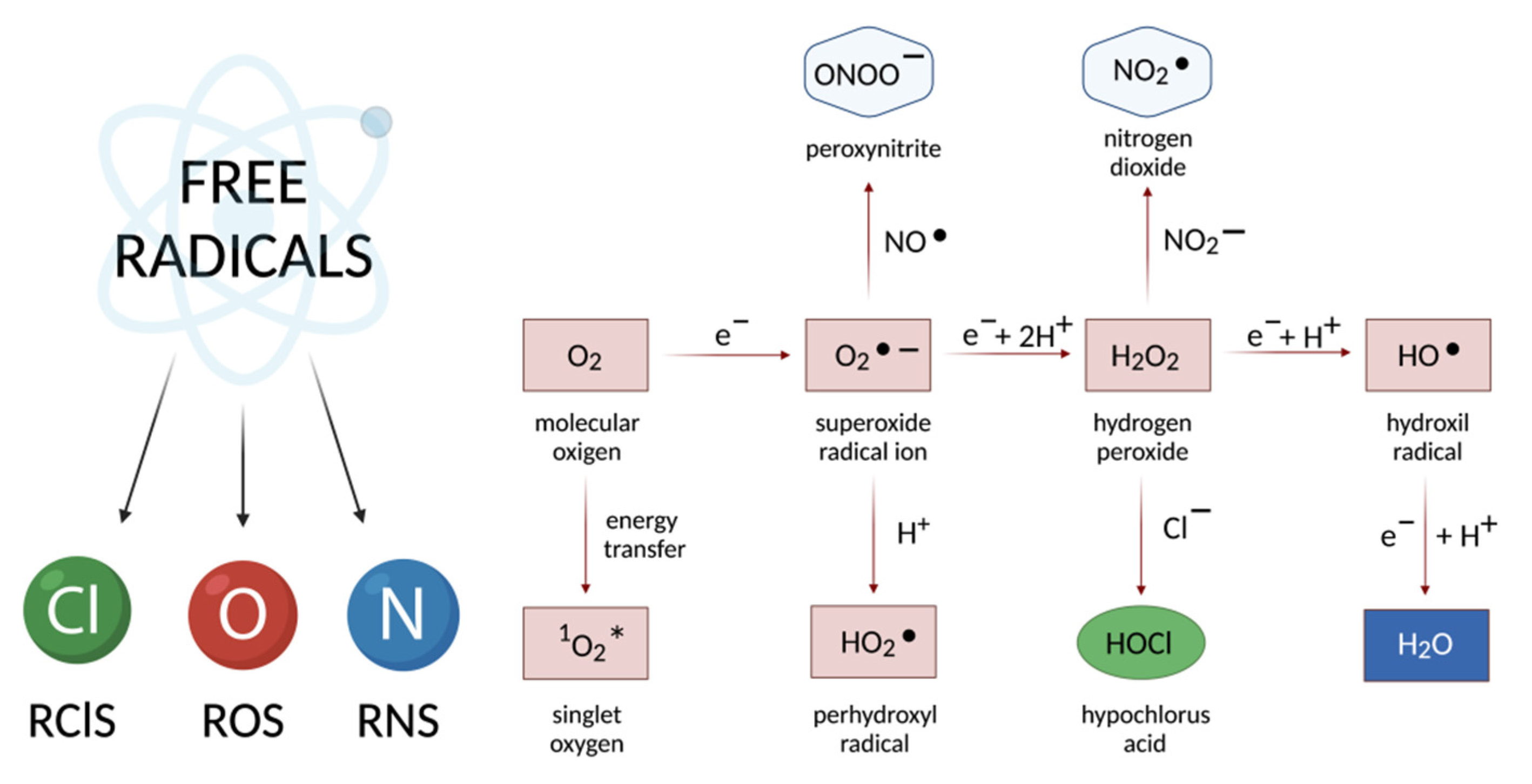
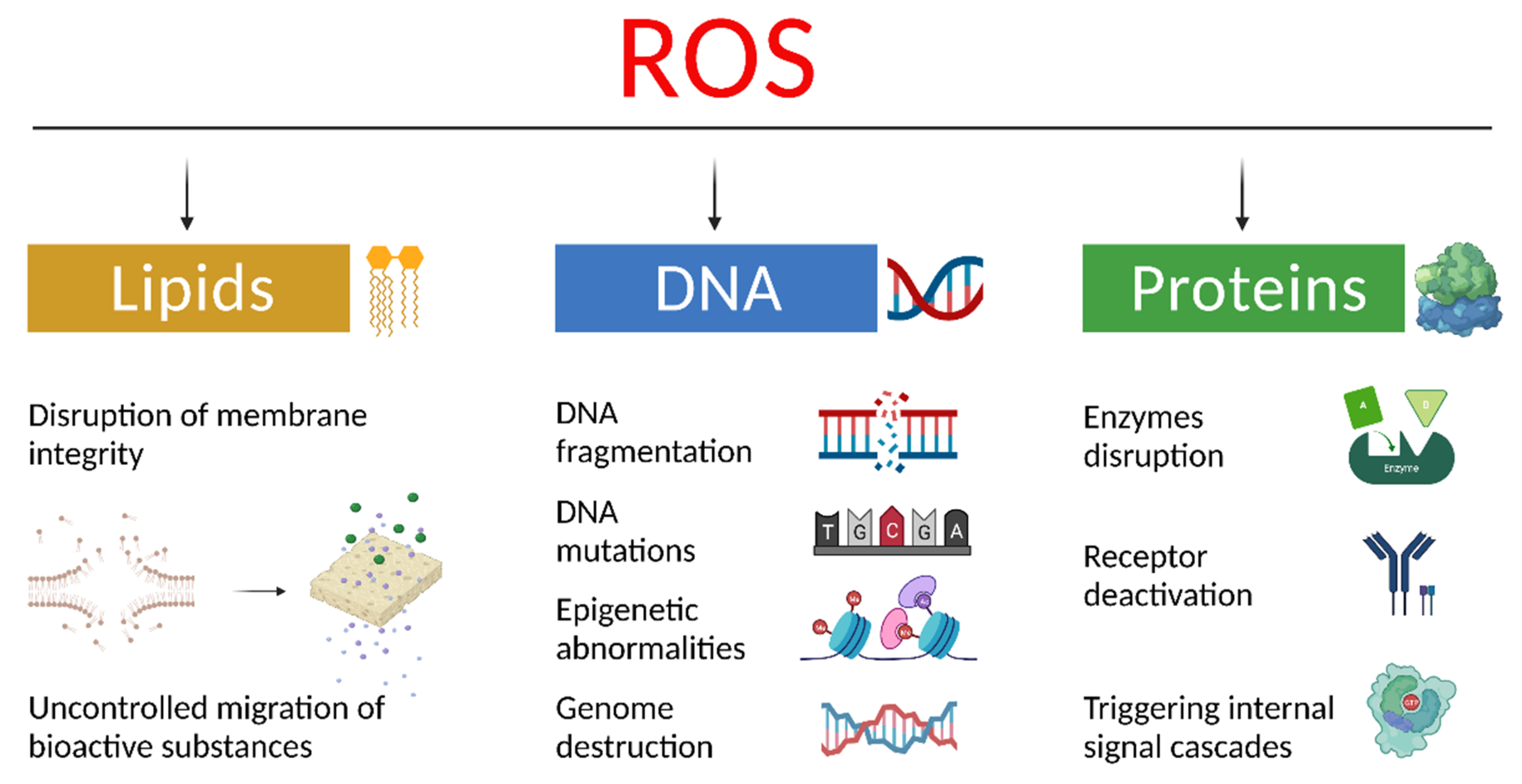

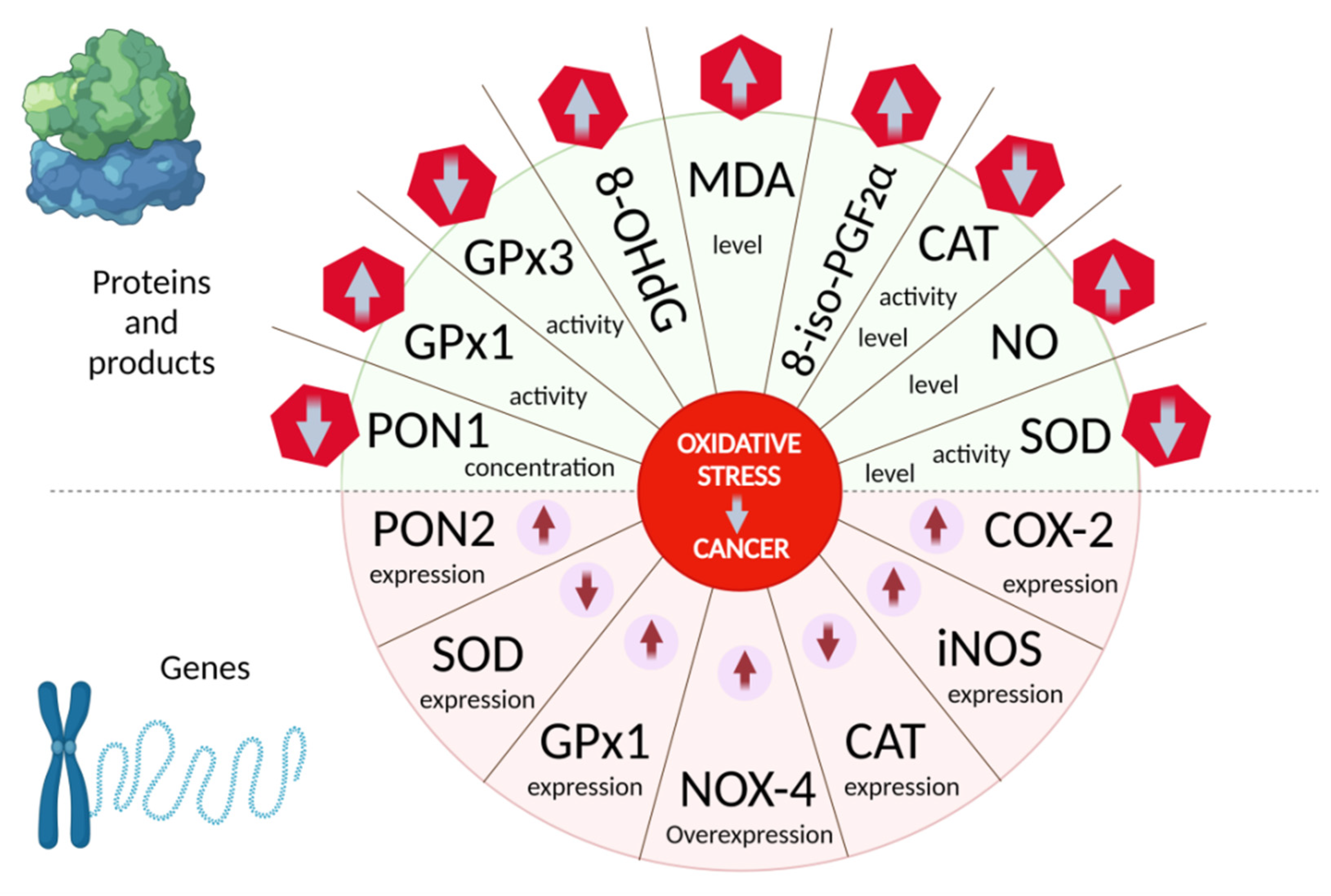






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Enzyme | Characteristics of the Enzyme | Ref. |
|---|---|---|---|
| COX-2 | cyclooxygenase 2 | COX-2 is a key enzyme in the biosynthesis of prostaglandins (mainly PGE2) and thromboxanes due to the conversion of arachidonic acid. In malignant neoplasms, COX-2 is an important regulator of angiogenesis, inflammation, and tumor formation, and plays an important role in metastasis, as evidenced by the high level of this enzyme in carcinogenesis. | [73,74,75,76,77,78,79,80] |
| NOX-4 | nicotinamide adenine dinucleotide phosphate oxidase subunit 4 | Nicotinamide adenine dinucleotide phosphate oxidase subunit 4 (NOX4) is an enzyme expressed by thyroid cells and regulates the production of reactive oxygen species (H2O2). Aberrant NOX4 expression contributes to a high rate of DNA mutagenesis and correlates with poor tumor prognosis and low patient survival. | [81,82] |
| iNOS | inducible nitric oxide synthase | Induced nitric oxide synthase is an enzyme responsible for the production of nitric oxide, which is absent in most cells under normal conditions. Aberrant induction of iNOS expression and activation accompanies all stages of carcinogenesis and is also associated with the development of drug resistance phenomenon, a high risk of relapse and death of patients. | [83,84,85] |
| CAT | catalase | Catalase is a key enzyme in the H2O2 metabolism and active nitrogen forms. Impairment of the expression and localization of this enzyme is characterized with tumor cells in numerous cancer types. | [86,87,88] |
| GPx3 | glutathione peroxidase 3 | Glutathione peroxidase 3 is a member of the selenoprotein family of glutathione peroxidase and participates in cell protection from oxidative damage ensuring the reduction of organic hydroperoxides and hydrogen peroxide via glutathione. Reduced levels of this enzyme expression are found in tumor samples obtained from patients with various types of malignant neoplasms, which indicate its function as a tumor suppressor. | [89,90,91] |
| SOD1 | superoxide dismutase | Superoxide dismutases are a class of enzymes that catalyze the conversion of superoxide radicals into oxygen and hydrogen peroxide. Copper SOD (Cu/ZnSOD, SOD1) is responsible for the regulation of superoxide levels in the intermembrane space of mitochondria, cytosol, and peroxisome, while manganese SOD (MnSOD, SOD2) is the main antioxidant enzyme that absorbs the superoxide radical anion in mitochondria. The enzymatic activity of superoxide dismutases is often reduced in the early cancer stages, while tumor cells contain low levels of SOD proteins. | [92,93,94,95] |
| PON | serum paraoxonase/ arylesterase | The family of antioxidant enzymes paraoxonases consists of three representatives: PON1, PON2, and PON3. The changes in PON status, covering the genotype, activity, and/or expression, were found in cancer patients, as well as in various tumor cell lines. The role of these enzymes in the survival of transformed cells and the formation of chemotherapeutic resistance is shown. | [96,97,98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neganova, M.; Liu, J.; Aleksandrova, Y.; Klochkov, S.; Fan, R. Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment. Cancers 2021, 13, 6062. https://doi.org/10.3390/cancers13236062
Neganova M, Liu J, Aleksandrova Y, Klochkov S, Fan R. Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment. Cancers. 2021; 13(23):6062. https://doi.org/10.3390/cancers13236062
Chicago/Turabian StyleNeganova, Margarita, Junqi Liu, Yulia Aleksandrova, Sergey Klochkov, and Ruitai Fan. 2021. "Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment" Cancers 13, no. 23: 6062. https://doi.org/10.3390/cancers13236062