
Lateralized and Segmental Overgrowth in Children

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
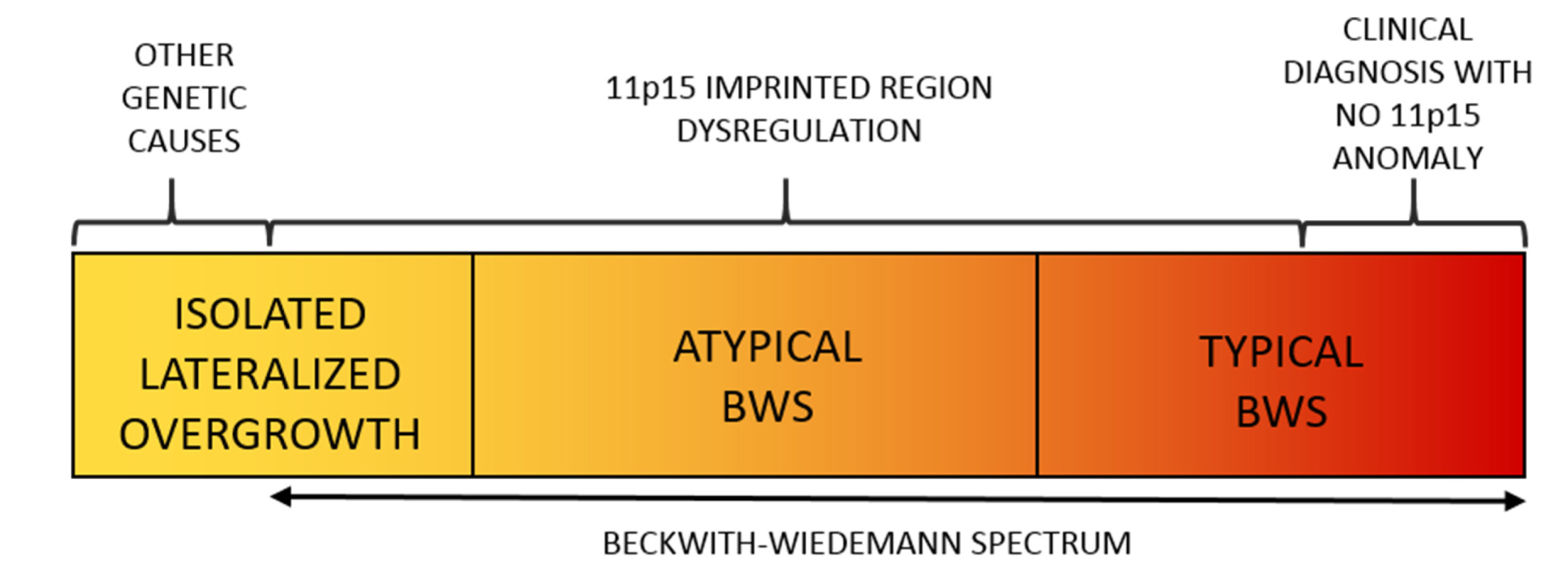
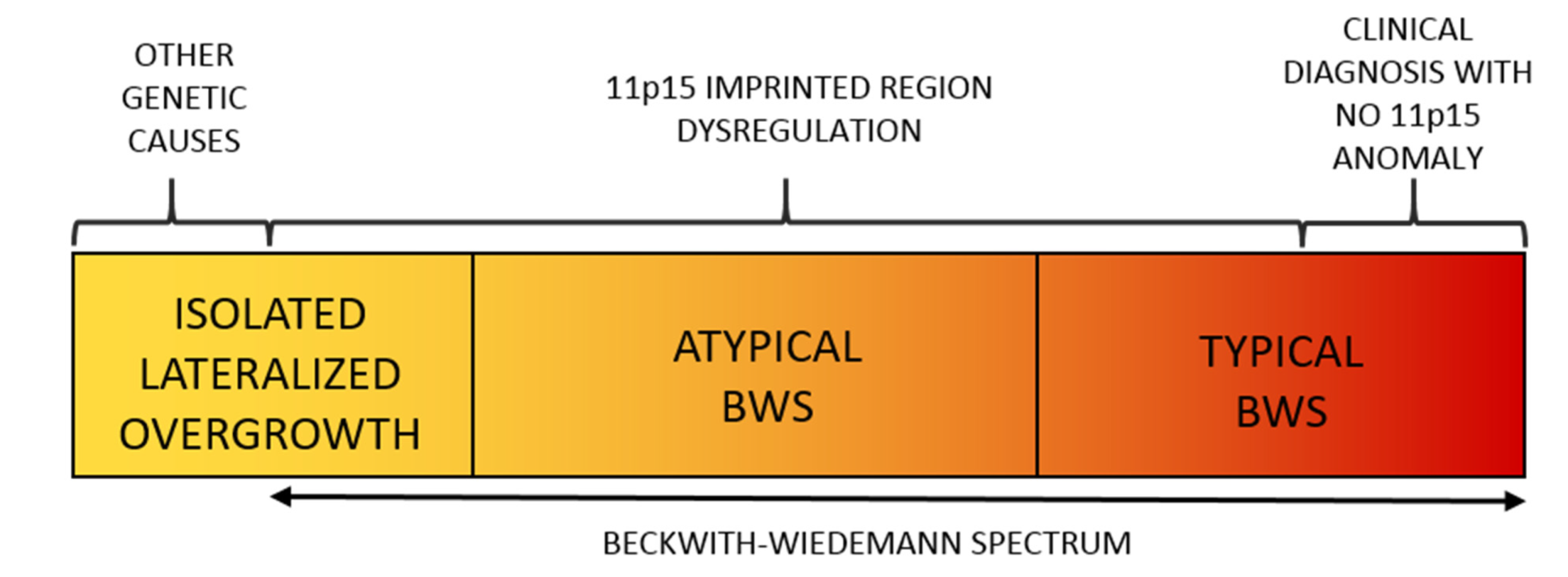
2. Beckwith–Wiedemann Spectrum (BWSp)
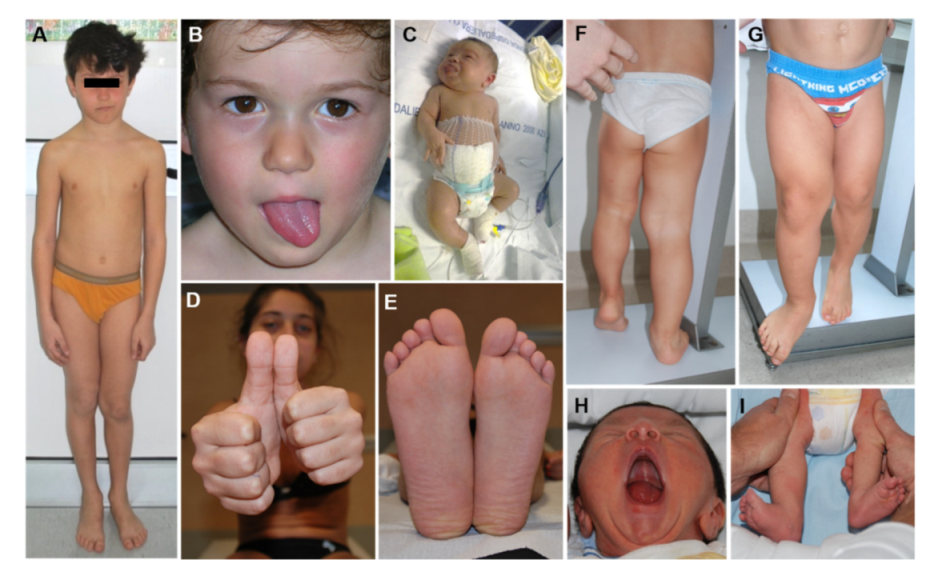
2.1. Overview
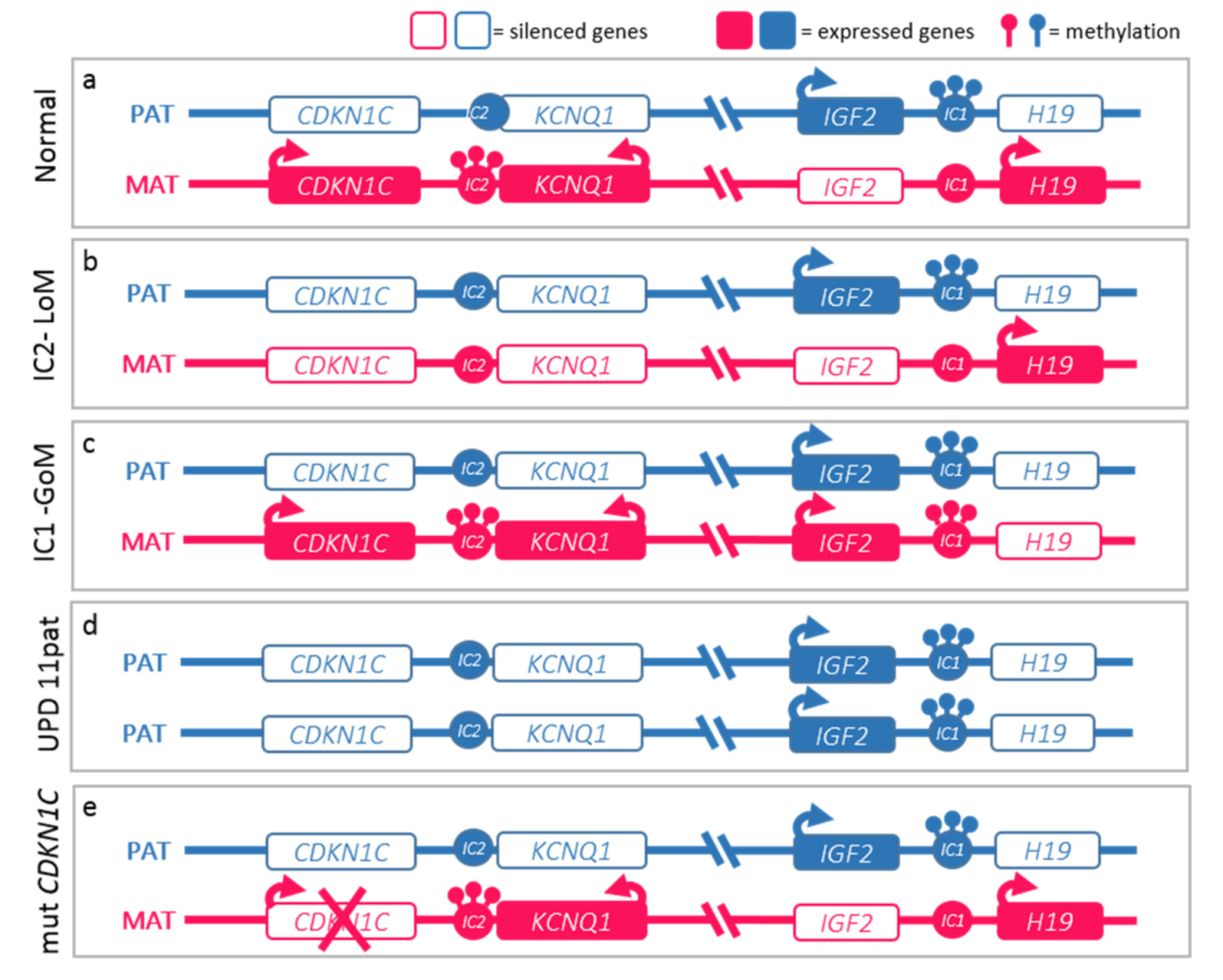
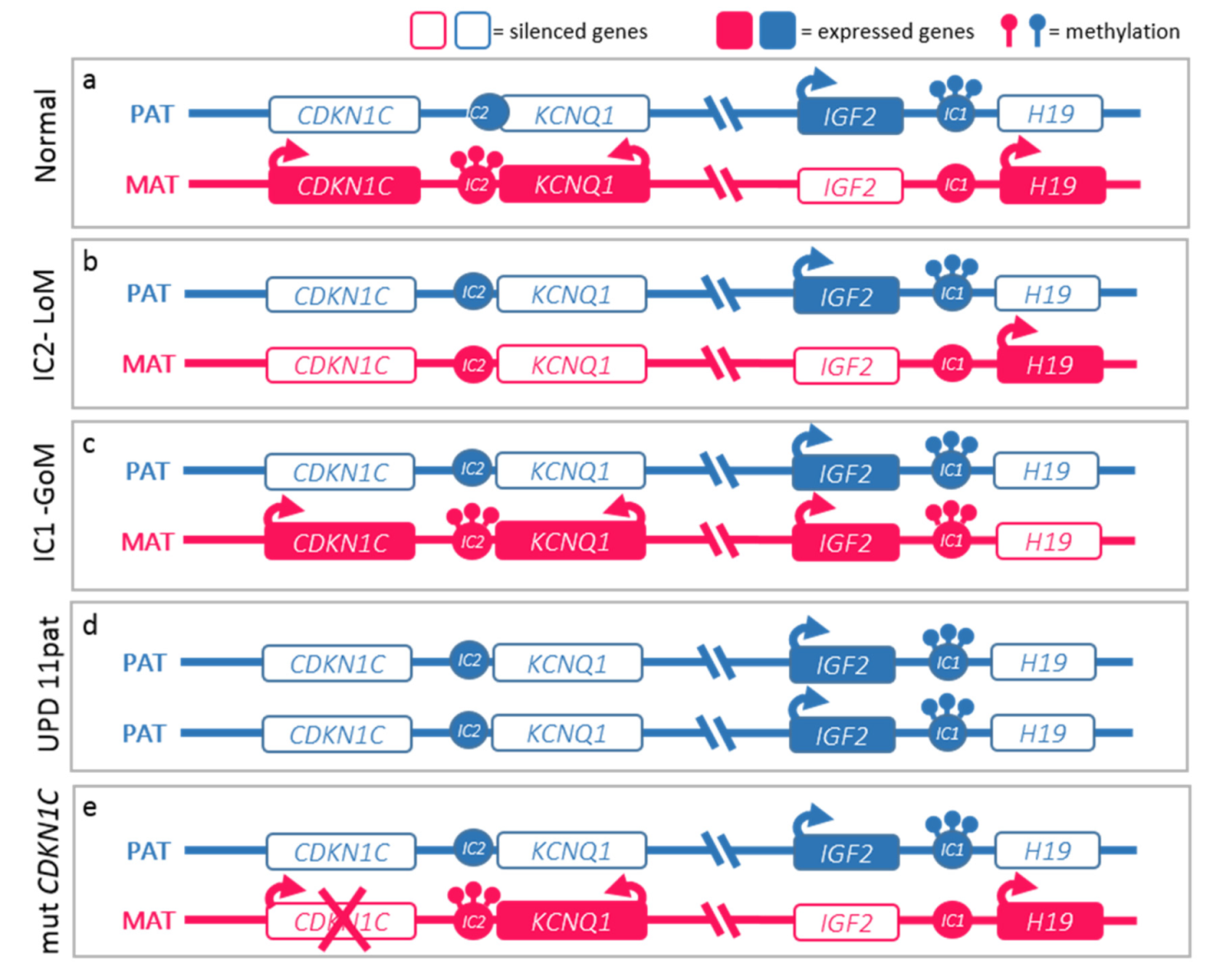
2.2. Molecular Basis of the BWS Spectrum
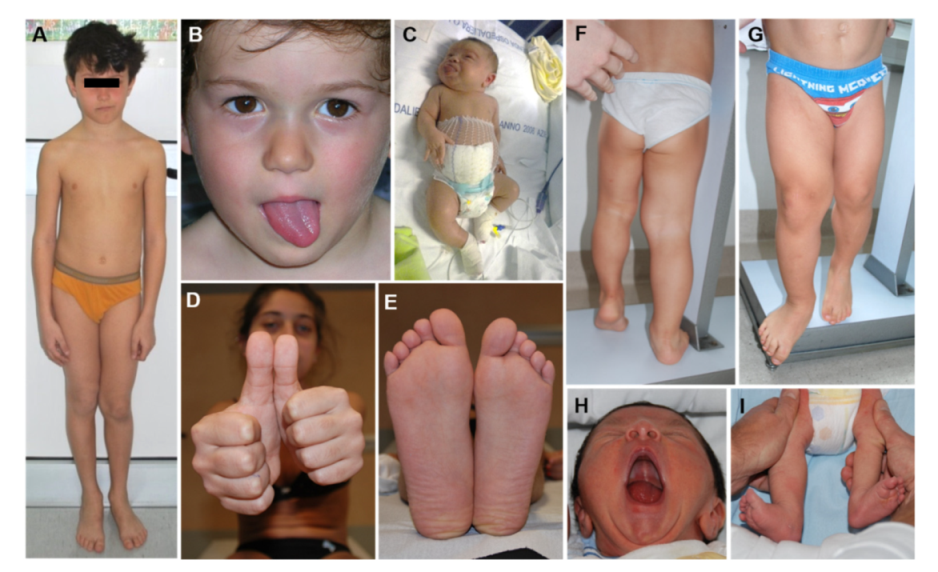
2.3. Lateralized Overgrowth in BWSp
2.4. Cancer Screening
3. PIK3CA-Related Overgrowth Spectrum (PROS)
3.1. Overview
- Type I macrodactyly [43];
- Muscle fibrous hyperplasia (MH, FH) [44] associated or not with vascular anomalies (fibroadipose vascular anomalies, FAVA) or adipose overgrowth (fibroadipose overgrowth, FAO) including hemihyperplasia multiple lipomatosis (HHML) characterized by overgrowth of the entire hemi-body and multiple lipomas;
- Facial Infiltrating Lipomatosis (FIL) is characterized by overgrowth mostly involving adipose and soft tissue localized to the facial district with a clear-cut asymmetry of the face;
- Epidermal nevi (EN), Seborrheic Keratosis (SK) and benign lichenoid keratosis (BLK);
- Congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal syndrome (CLOVES, OMIM # 612918) with or without acral deformities as large, wide feet and hands, macrodactyly, and sandal gap [45];
- Klippel–Trenaunay syndrome (KTS, OMIM # 149000) is characterized by disproportionate growth of soft and bony tissue of a body region (typically involving the lower extremities) and combined with cutaneous capillary, lymphatic, and venous malformations consisting in varicosity, the persistence of embryonal veins, or a lymphatic component [46];
- Megalencephaly-capillary malformation polymicrogyria syndrome (OMIM # 602501, MCAP), characterized by macrocephaly, capillary malformation, somatic overgrowth, body asymmetry, brain anomalies and progressive megalencephaly, and neurodevelopmental delay [47];
- Hemimegalencephaly (H-MEG) and dysplastic megalencephaly (D-MEG) characterized by cerebral overgrowth and a variable degree of cortical development malformation [48];
- Capillary malformation of the lower lip, lymphatic malformation of the face and neck, asymmetry and partial/generalized overgrowth (CLAPO, OMIM # 613089) [49].
- Finding of somatic mutation in the PIK3CA gene;
- Congenital or early childhood clinical presentation;
- Localized growth trend with a mosaic pattern;
- Presence of at least a or b criteria:
- (a)
- Two or three of the following anomalies:
- i.
- Overgrowth: adipose, muscular, nervous, skeletal
- ii.
- Vascular malformations: capillary, venous, arteriovenous, lymphatic
- iii.
- Epidermal nevus
- (b)
- Presence of at least one of the following isolated anomalies:
- iv.
- Extensive lymphatic malformation
- v.
- Macrodactyly or acral overgrowth (hands, feet, limb)
- vi.
- Fat overgrowth localized to the trunk
- vii.
- Hemimegalencephaly/dysplastic megalencephaly/focal cortical dysplasia
- viii.
- Epidermal nevus
- ix.
- Seborrheic keratosis
- x.
- Benign lichenoid keratosis
3.2. Molecular Diagnosis
3.3. Pathophysiology and Druggability
3.4. PROS and Cancer
4. Proteus Syndrome and AKT1-Related Overgrowth Spectrum
5. PTEN Hamartoma Tumor Syndrome (PHTS)
6. Mosaic RASopathies
7. Lateralized Overgrowth Syndromes with Vascular Anomalies
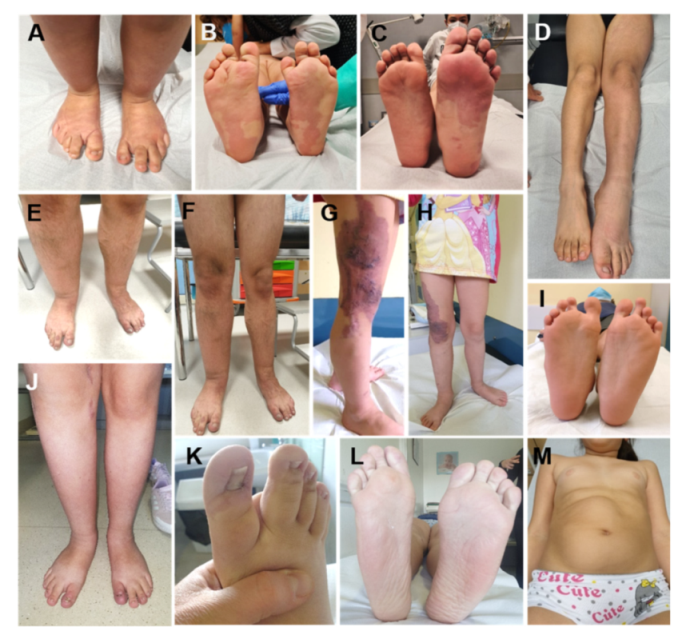
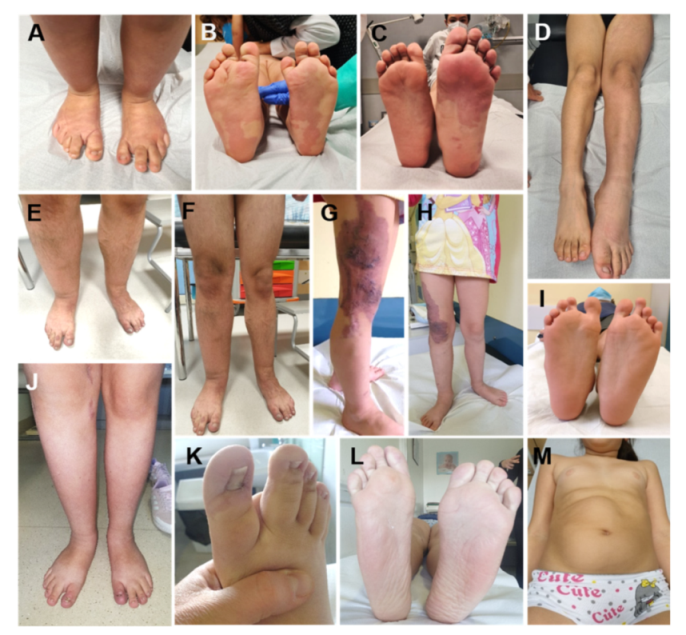
8. Isolated Lateralized Overgrowth (Not Belonging to Other Overgrowth Disorders)
8.1. Overview
8.2. Molecular Bases
8.3. Cancer Risk and Surveillance
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalish, J.M.; Biesecker, L.G.; Brioude, F.; Deardorff, M.A.; Di Cesare-Merlone, A.; Druley, T.; Ferrero, G.B.; Lapunzina, P.; Larizza, L.; Maas, S.; et al. Nomenclature and definition in asymmetric regional body overgrowth. Am. J. Med. Genet. A 2017, 173, 1735–1738. [Google Scholar] [CrossRef]
- Lacerda, L.D.S.; Alves, U.D.; Zanier, J.F.; Machado, D.C.; Camilo, G.B.; Lopes, A.J. Differential diagnoses of overgrowth syndromes: The most important clinical and radiological disease manifestations. Radiol. Res. Pract. 2014, 2014, 947451. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Suttie, M.; Bulstrode, N.W.; Britto, J.A.; Dunaway, D.; Hammond, P.; Ferretti, P. Combined soft and skeletal tissue modelling of normal and dysmorphic midface postnatal development. J. Craniomaxillofac. Surg. 2016, 44, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Griff, J.R.; Duffy, K.A.; Kalish, J.M. Characterization and Childhood Tumor Risk Assessment of Genetic and Epigenetic Syndromes Associated with Lateralized Overgrowth. Front. Pediatr. 2020, 8, 613260. [Google Scholar] [CrossRef]
- Manor, J.; Lalani, S.R. Overgrowth Syndromes-Evaluation, Diagnosis, and Management. Front. Pediatr. 2020, 8, 574857. [Google Scholar] [CrossRef]
- Mussa, A.; Russo, S.; De Crescenzo, A.; Chiesa, N.; Molinatto, C.; Selicorni, A.; Richiardi, L.; Larizza, L.; Silengo, M.C.; Riccio, A.; et al. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am. J. Med. Genet. A 2013, 161, 2481–2486. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Cerrato, F.; Palumbo, O.; Carella, M.; Baldassarre, G.; Carli, D.; Peris, C.; Riccio, A.; Ferrero, G.B. Assisted Reproductive Techniques and Risk of Beckwith-Wiedemann Syndrome. Pediatrics 2017, 140, e20164311. [Google Scholar] [CrossRef] [Green Version]
- Carli, D.; Bertola, C.; Cardaropoli, S.; Ciuffreda, V.P.; Pieretto, M.; Ferrero, G.B.; Mussa, A. Prenatal features in Beckwith-Wiedemann syndrome and indications for prenatal testing. J. Med. Genet. 2020, 58, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Ferrero, G.B.; Ceoloni, B.; Basso, E.; Chiesa, N.; De Crescenzo, A.; Pepe, E.; Silengo, M.; de Sanctis, L. Neonatal hepatoblastoma in a newborn with severe phenotype of Beckwith-Wiedemann syndrome. Eur. J. Pediatr. 2011, 170, 1407–1411. [Google Scholar] [CrossRef]
- Baker, S.W.; Ryan, E.; Kalish, J.M.; Ganguly, A. Prenatal molecular testing and diagnosis of Beckwith-Wiedemann syndrome. Prenat. Diagn. 2021, 41, 817–822. [Google Scholar] [CrossRef]
- Mussa, A.; Russo, S.; de Crescenzo, A.; Freschi, A.; Calzari, L.; Maitz, S.; Macchiaiolo, M.; Molinatto, C.; Baldassarre, G.; Mariani, M.; et al. Fetal growth patterns in Beckwith-Wiedemann syndrome. Clin. Genet. 2016, 90, 21–27. [Google Scholar] [CrossRef]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Duffy, K.A.; Cielo, C.M.; Cohen, J.L.; Gonzalez-Gandolfi, C.X.; Griff, J.R.; Hathaway, E.R.; Kupa, J.; Taylor, J.A.; Wang, K.H.; Ganguly, A.; et al. Characterization of the Beckwith-Wiedemann spectrum: Diagnosis and management. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 693–708. [Google Scholar] [CrossRef]
- Shuman, C.; Beckwith, J.B.; Weksberg, R. Beckwith-Wiedemann Syndrome. In GeneReviews (®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Mussa, A.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: A paradigm for genomic medicine. Clin. Genet. 2016, 89, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Russo, S.; De Crescenzo, A.; Freschi, A.; Calzari, L.; Maitz, S.; Macchiaiolo, M.; Molinatto, C.; Baldassarre, G.; Mariani, M.; et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2016, 24, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Cooper, W.N.; Luharia, A.; Evans, G.A.; Raza, H.; Haire, A.C.; Grundy, R.; Bowdin, S.C.; Riccio, A.; Sebastio, G.; Bliek, J.; et al. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2005, 13, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Tucker, M.A. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J. Pediatr. 1998, 132, 398–400. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Kirby, G.; Hardy, C.; Dias, R.P.; Tee, L.; Lim, D.; Berg, J.; MacDonald, F.; Nightingale, P.; Maher, E.R. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin. Epigenet. 2014, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porteus, M.H.; Narkool, P.; Neuberg, D.; Guthrie, K.; Breslow, N.; Green, D.M.; Diller, L. Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms’ tumor: A report from the National Wilms Tumor Study Group. J. Clin. Oncol. 2000, 18, 2026–2031. [Google Scholar] [CrossRef]
- Carli, D.; De Pellegrin, M.; Franceschi, L.; Zinali, F.; Paonessa, M.; Spolaore, S.; Cardaropoli, S.; Cravino, M.; Marcucci, L.; Andreacchio, A.; et al. Evolution over Time of Leg Length Discrepancy in Patients with Syndromic and Isolated Lateralized Overgrowth. J. Pediatr. 2021, 234, 123–127. [Google Scholar] [CrossRef]
- Gazzin, A.; Carli, D.; Sirchia, F.; Molinatto, C.; Cardaropoli, S.; Palumbo, G.; Zampino, G.; Ferrero, G.B.; Mussa, A. Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2019, 179, 1691–1702. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Baldassarre, G.; Riberi, E.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J. Pediatr. 2016, 176, 142.e1–149.e1. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.M.; Vansenne, F.; Kadouch, D.J.; Ibrahim, A.; Bliek, J.; Hopman, S.; Mannens, M.M.; Merks, J.H.; Maher, E.R.; Hennekam, R.C. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am. J. Med. Genet. A 2016, 170, 2248–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussa, A.; Duffy, K.A.; Carli, D.; Griff, J.R.; Fagiano, R.; Kupa, J.; Brodeur, G.M.; Ferrero, G.B.; Kalish, J.M. The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J. Cancer Res. Clin. Oncol. 2019, 145, 3115–3123. [Google Scholar] [CrossRef]
- Mussa, A.; Ciuffreda, V.P.; Sauro, P.; Pagliardini, V.; Pagliardini, S.; Carli, D.; Kalish, J.M.; Fagioli, F.; Pavanello, E.; Ferrero, G.B. Longitudinal Monitoring of Alpha-Fetoprotein by Dried Blood Spot for Hepatoblastoma Screening in Beckwith⁻Wiedemann Syndrome. Cancers 2019, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussa, A.; Pagliardini, S.; Pagliardini, V.; Molinatto, C.; Baldassarre, G.; Corrias, A.; Silengo, M.C.; Ferrero, G.B. α-Fetoprotein assay on dried blood spot for hepatoblastoma screening in children with overgrowth-cancer predisposition syndromes. Pediatr. Res. 2014, 76, 544–548. [Google Scholar] [CrossRef] [Green Version]
- Duffy, K.A.; Cohen, J.L.; Elci, O.U.; Kalish, J.M. Development of the Serum α-Fetoprotein Reference Range in Patients with Beckwith-Wiedemann Spectrum. J. Pediatr. 2019, 212, 195.e2–200.e2. [Google Scholar] [CrossRef] [PubMed]
- Clericuzio, C.L.; Chen, E.; McNeil, D.E.; O’Connor, T.; Zackai, E.H.; Medne, L.; Tomlinson, G.; DeBaun, M. Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J. Pediatr. 2003, 143, 270–272. [Google Scholar] [CrossRef]
- Mussa, A.; Ferrero, G.B. Serum alpha-fetoprotein screening for hepatoblastoma in Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2017, 173, 585–587. [Google Scholar] [CrossRef]
- Mussa, A.; Ferrero, G.B. Screening Hepatoblastoma in Beckwith-Wiedemann Syndrome: A Complex Issue. J. Pediatr. Hematol. Oncol. 2015, 37, 627. [Google Scholar] [CrossRef]
- Kalish, J.M.; Deardorff, M.A. Tumor screening in Beckwith-Wiedemann syndrome-To screen or not to screen? Am. J. Med. Genet. A 2016, 170, 2261–2264. [Google Scholar] [CrossRef] [Green Version]
- Kalish, J.M.; Doros, L.; Helman, L.J.; Hennekam, R.C.; Kuiper, R.P.; Maas, S.M.; Maher, E.R.; Nichols, K.E.; Plon, S.E.; Porter, C.C.; et al. Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin. Cancer Res. 2017, 23, e115–e122. [Google Scholar] [CrossRef] [Green Version]
- Mussa, A.; Duffy, K.A.; Carli, D.; Ferrero, G.B.; Kalish, J.M. Defining an optimal time window to screen for hepatoblastoma in children with Beckwith-Wiedemann syndrome. Pediatr. Blood Cancer 2019, 66, e27492. [Google Scholar] [CrossRef]
- Sheppard, S.E.; Lalonde, E.; Adzick, N.S.; Beck, A.E.; Bhatti, T.; De Leon, D.D.; Duffy, K.A.; Ganguly, A.; Hathaway, E.; Ji, J.; et al. Androgenetic chimerism as an etiology for Beckwith-Wiedemann syndrome: Diagnosis and management. Genet. Med. 2019, 21, 2644–2649. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Mostoufi-Moab, S.; Zelley, K.; Mattei, P.A.; States, L.J.; Bhatti, T.R.; Duffy, K.A.; Brodeur, G.M.; Kalish, J.M. Management of adrenal masses in patients with Beckwith-Wiedemann syndrome. Pediatr. Blood Cancer 2017, 64, e26432. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Duffy, K.A.; Bhatti, T.R.; Bagatell, R.; Balamuth, N.J.; Brodeur, G.M.; Ganguly, A.; Mattei, P.A.; Surrey, L.F.; Balis, F.M.; et al. Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatr. Blood Cancer 2018, 65, e27296. [Google Scholar] [CrossRef]
- Fiala, E.M.; Ortiz, M.V.; Kennedy, J.A.; Glodzik, D.; Fleischut, M.H.; Duffy, K.A.; Hathaway, E.R.; Heaton, T.; Gerstle, J.T.; Steinherz, P.; et al. 11p15.5 epimutations in children with Wilms tumor and hepatoblastoma detected in peripheral blood. Cancer 2020, 126, 3114–3121. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Rios, J.J.; Parker, V.E.; Semple, R.K.; Lindhurst, M.J.; Sapp, J.C.; Alomari, A.; Ezaki, M.; Dobyns, W.; Biesecker, L.G. PIK3CA-related overgrowth spectrum (PROS): Diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am. J. Med. Genet. A 2015, 167A, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Mirzaa, G.; Conway, R.; Graham, J.M., Jr.; Dobyns, W.B. PIK3CA-Related Segmental Overgrowth. In GeneReviews (®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chang, F.; Liu, L.; Fang, E.; Zhang, G.; Chen, T.; Cao, K.; Li, Y.; Li, M.M. Molecular Diagnosis of Mosaic Overgrowth Syndromes Using a Custom-Designed Next-Generation Sequencing Panel. J. Mol. Diagn. 2017, 19, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.; Hao, M.; Luu, M. PIK3CA vascular overgrowth syndromes: An update. Curr. Opin. Pediatr. 2020, 32, 539–546. [Google Scholar] [CrossRef]
- Labow, B.I.; Pike, C.M.; Upton, J. Overgrowth of the Hand and Upper Extremity and Associated Syndromes. J. Hand Surg. Am. 2016, 41, 473–482. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Parker, V.E.; Payne, F.; Sapp, J.C.; Rudge, S.; Harris, J.; Witkowski, A.M.; Zhang, Q.; Groeneveld, M.P.; Scott, C.E.; et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat. Genet. 2012, 44, 928–933. [Google Scholar] [CrossRef]
- Alomari, A.I. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: A descriptive study of 18 cases of CLOVES syndrome. Clin. Dysmorphol. 2009, 18, 1–7. [Google Scholar] [CrossRef]
- Oduber, C.E.; van der Horst, C.M.; Hennekam, R.C. Klippel-Trenaunay syndrome: Diagnostic criteria and hypothesis on etiology. Ann. Plast. Surg. 2008, 60, 217–223. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; Conway, R.L.; Gripp, K.W.; Lerman-Sagie, T.; Siegel, D.H.; deVries, L.S.; Lev, D.; Kramer, N.; Hopkins, E.; Graham, J.M.; et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: Two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am. J. Med. Genet. A 2012, 158A, 269–291. [Google Scholar] [CrossRef]
- Dobyns, W.B.; Mirzaa, G.M. Megalencephaly syndromes associated with mutations of core components of the PI3K-AKT-MTOR pathway: PIK3CA, PIK3R2, AKT3, and MTOR. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 582–590. [Google Scholar] [CrossRef]
- López-Gutiérrez, J.C.; Lapunzina, P. Capillary malformation of the lower lip, lymphatic malformation of the face and neck, asymmetry and partial/generalized overgrowth (CLAPO): Report of six cases of a new syndrome/association. Am. J. Med. Genet. A 2008, 146A, 2583–2588. [Google Scholar] [CrossRef]
- Madsen, R.R.; Vanhaesebroeck, B.; Semple, R.K. Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends Mol. Med. 2018, 24, 856–870. [Google Scholar] [CrossRef] [Green Version]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Castillo, S.D.; Tzouanacou, E.; Zaw-Thin, M.; Berenjeno, I.M.; Parker, V.E.; Chivite, I.; Milà-Guasch, M.; Pearce, W.; Solomon, I.; Angulo-Urarte, A.; et al. Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci. Transl. Med. 2016, 8, 332ra43. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Skibo, J.; Kalume, F.; Ni, J.; Rankin, S.; Lu, Y.; Dobyns, W.B.; Mills, G.B.; Zhao, J.J.; Baker, S.J.; et al. Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy. eLife 2015, 4, e12703. [Google Scholar] [CrossRef]
- Madsen, R.R. PI3K in stemness regulation: From development to cancer. Biochem. Soc. Trans. 2020, 48, 301–315. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Parker, V.E.R.; Keppler-Noreuil, K.M.; Faivre, L.; Luu, M.; Oden, N.L.; De Silva, L.; Sapp, J.C.; Andrews, K.; Bardou, M.; Chen, K.Y.; et al. Safety and efficacy of low-dose sirolimus in the PIK3CA-related overgrowth spectrum. Genet. Med. 2019, 21, 1189–1198. [Google Scholar] [CrossRef] [Green Version]
- Forde, K.; Resta, N.; Ranieri, C.; Rea, D.; Kubassova, O.; Hinton, M.; Andrews, K.A.; Semple, R.; Irvine, A.D.; Dvorakova, V. Clinical experience with the AKT1 inhibitor miransertib in two children with PIK3CA-related overgrowth syndrome. Orphanet J. Rare Dis. 2021, 16, 109. [Google Scholar] [CrossRef]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef] [PubMed]
- Pagliazzi, A.; Oranges, T.; Traficante, G.; Trapani, C.; Facchini, F.; Martin, A.; Semeraro, A.; Perrone, A.; Filippeschi, C.; Giglio, S. PIK3CA-Related Overgrowth Spectrum from Diagnosis to Targted Therapy: A Case of CLOVES Syndrome Treated with Alpelisib. Front. Pediatr. 2021, 9, 732836. [Google Scholar] [CrossRef]
- Venot, Q.; Blanc, T.; Rabia, S.H.; Berteloot, L.; Ladraa, S.; Duong, J.P.; Blanc, E.; Johnson, S.C.; Hoguin, C.; Boccara, O.; et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018, 558, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Hammill, A.M.; Adams, D.; Vikkula, M.; Keppler-Noreuil, K.M. A review of mechanisms of disease across PIK3CA-related disorders with vascular manifestations. Orphanet J. Rare Dis. 2021, 16, 306. [Google Scholar] [CrossRef]
- Peterman, C.M.; Fevurly, R.D.; Alomari, A.I.; Trenor, C.C.; Adams, D.M.; Vadeboncoeur, S.; Liang, M.G.; Greene, A.K.; Mulliken, J.B.; Fishman, S.J. Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr. Blood Cancer 2017, 64, e26684. [Google Scholar] [CrossRef]
- Sapp, J.C.; Buser, A.; Burton-Akright, J.; Keppler-Noreuil, K.M.; Biesecker, L.G. A dyadic genotype-phenotype approach to diagnostic criteria for Proteus syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 565–570. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Sapp, J.C.; Teer, J.K.; Johnston, J.J.; Finn, E.M.; Peters, K.; Turner, J.; Cannons, J.L.; Bick, D.; Blakemore, L.; et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med. 2011, 365, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Buser, A.; Lindhurst, M.J.; Kondolf, H.C.; Yourick, M.R.; Keppler-Noreuil, K.M.; Sapp, J.C.; Biesecker, L.G. Allelic heterogeneity of Proteus syndrome. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005181. [Google Scholar] [CrossRef]
- Leoni, C.; Gullo, G.; Resta, N.; Fagotti, A.; Onesimo, R.; Schwartz, B.; Kazakin, J.; Abbadessa, G.; Crown, J.; Collins, C.D.; et al. First evidence of a therapeutic effect of miransertib in a teenager with Proteus syndrome and ovarian carcinoma. Am. J. Med. Genet. A 2019, 179, 1319–1324. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Sapp, J.C.; Lindhurst, M.J.; Darling, T.N.; Burton-Akright, J.; Bagheri, M.; Dombi, E.; Gruber, A.; Jarosinski, P.F.; Martin, S.; et al. Pharmacodynamic Study of Miransertib in Individuals with Proteus Syndrome. Am. J. Hum. Genet. 2019, 104, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehia, L.; Eng, C. PTEN Hamartoma Tumor Syndrome. In GeneReviews (®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 1993; Copyright © 1993–2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Wash-ington, Seattle. All rights reserved. [Google Scholar]
- Caux, F.; Plauchu, H.; Chibon, F.; Faivre, L.; Fain, O.; Vabres, P.; Bonnet, F.; Selma, Z.B.; Laroche, L.; Gérard, M.; et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur. J. Hum. Genet. 2007, 15, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.P.; Marsh, D.J.; Hampel, H.; Mulliken, J.B.; Gimm, O.; Eng, C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum. Mol. Genet. 2000, 9, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Nathan, N.; Keppler-Noreuil, K.M.; Biesecker, L.G.; Moss, J.; Darling, T.N. Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol. Clin. 2017, 35, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Mester, J.; Eng, C. When overgrowth bumps into cancer: The PTEN-opathies. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 114–121. [Google Scholar] [CrossRef]
- Simpson, L.; Parsons, R. PTEN: Life as a tumor suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Jonker, L.A.; Lebbink, C.A.; Jongmans, M.C.J.; Nievelstein, R.A.J.; Merks, J.H.M.; Nieveen van Dijkum, E.J.M.; Links, T.P.; Hoogerbrugge, N.; van Trotsenburg, A.S.P.; van Santen, H.M. Recommendations on Surveillance for Differentiated Thyroid Carcinoma in Children with PTEN Hamartoma Tumor Syndrome. Eur. Thyroid J. 2020, 9, 234–242. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; PHTS Guideline Development Group; The European Reference Network GENTURIS. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur. J. Hum. Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef]
- Komiya, T.; Blumenthal, G.M.; DeChowdhury, R.; Fioravanti, S.; Ballas, M.S.; Morris, J.; Hornyak, T.J.; Wank, S.; Hewitt, S.M.; Morrow, B.; et al. A Pilot Study of Sirolimus in Subjects with Cowden Syndrome or Other Syndromes Characterized by Germline Mutations in PTEN. Oncologist 2019, 24, 1510-e1265. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, I.; Burrows, P.E.; Adams, D.M.; Sutton, V.R.; Hollier, L.H.; Chintagumpala, M.M. Oral rapamycin in the treatment of patients with hamartoma syndromes and PTEN mutation. Pediatr. Blood Cancer 2011, 57, 321–323. [Google Scholar] [CrossRef]
- Hardan, A.Y.; Jo, B.; Frazier, T.W.; Klaas, P.; Busch, R.M.; Dies, K.A.; Filip-Dhima, R.; Snow, A.V.; Eng, C.; Hanna, R.; et al. A randomized double-blind controlled trial of everolimus in individuals with PTEN mutations: Study design and statistical considerations. Contemp. Clin. Trials Commun. 2021, 21, 100733. [Google Scholar] [CrossRef]
- Boppudi, S.; Bögershausen, N.; Hove, H.B.; Percin, E.F.; Aslan, D.; Dvorsky, R.; Kayhan, G.; Li, Y.; Cursiefen, C.; Tantcheva-Poor, I.; et al. Specific mosaic KRAS mutations affecting codon 146 cause oculoectodermal syndrome and encephalocraniocutaneous lipomatosis. Clin. Genet. 2016, 90, 334–342. [Google Scholar] [CrossRef]
- Chacon-Camacho, O.F.; Lopez-Moreno, D.; Morales-Sanchez, M.A.; Hofmann, E.; Pacheco-Quito, M.; Wieland, I.; Cortes-Gonzalez, V.; Villanueva-Mendoza, C.; Zenker, M.; Zenteno, J.C. Expansion of the phenotypic spectrum and description of molecular findings in a cohort of patients with oculocutaneous mosaic RASopathies. Mol. Genet. Genom. Med. 2019, 7, e625. [Google Scholar] [CrossRef]
- Schmidt, V.F.; Wieland, I.; Wohlgemuth, W.A.; Ricke, J.; Wildgruber, M.; Zenker, M. Mosaic RASopathy due to KRAS variant G12D with segmental overgrowth and associated peripheral vascular malformations. Am. J. Med. Genet. A 2021, 185, 3122–3128. [Google Scholar] [CrossRef]
- Chang, C.A.; Perrier, R.; Kurek, K.C.; Estrada-Veras, J.; Lehman, A.; Yip, S.; Hendson, G.; Diamond, C.; Pinchot, J.W.; Tran, J.M.; et al. Novel findings and expansion of phenotype in a mosaic RASopathy caused by somatic KRAS variants. Am. J. Med. Genet. A 2021, 185, 2829–2845. [Google Scholar] [CrossRef] [PubMed]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Liang, M.G.; Mulliken, J.B. Diffuse capillary malformation with overgrowth: A clinical subtype of vascular anomalies with hypertrophy. J. Am. Acad. Dermatol. 2013, 69, 589–594. [Google Scholar] [CrossRef]
- Jordan, M.; Carmignac, V.; Sorlin, A.; Kuentz, P.; Albuisson, J.; Borradori, L.; Bourrat, E.; Boute, O.; Bukvic, N.; Bursztejn, A.C.; et al. Reverse Phenotyping in Patients with Skin Capillary Malformations and Mosaic GNAQ or GNA11 Mutations Defines a Clinical Spectrum with Genotype-Phenotype Correlation. J. Investig. Dermatol. 2020, 140, 1106.e2–1110.e2. [Google Scholar] [CrossRef]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Eggermann, T. Silver-Russell and Beckwith-Wiedemann syndromes: Opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm. Res. 2009, 71 (Suppl. S2), 30–35. [Google Scholar] [CrossRef]
- Zeschnigk, M.; Albrecht, B.; Buiting, K.; Kanber, D.; Eggermann, T.; Binder, G.; Gromoll, J.; Prott, E.C.; Seland, S.; Horsthemke, B. IGF2/H19 hypomethylation in Silver-Russell syndrome and isolated hemihypoplasia. Eur. J. Hum. Genet. 2008, 16, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Radley, J.A.; Connolly, M.; Sabir, A.; Kanani, F.; Carley, H.; Jones, R.L.; Hyder, Z.; Gompertz, L.; Reardon, W.; Richardson, R.; et al. Isolated- and Beckwith-Wiedemann syndrome related-lateralised overgrowth (hemihypertrophy): Clinical and molecular correlations in 94 individuals. Clin. Genet. 2021, 100, 292–297. [Google Scholar] [CrossRef]
- Dempsey-Robertson, M.; Wilkes, D.; Stall, A.; Bush, P. Incidence of abdominal tumors in syndromic and idiopathic hemihypertrophy/isolated hemihyperplasia. J. Pediatr. Orthop. 2012, 32, 322–326. [Google Scholar] [CrossRef]
- Dumoucel, S.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Parisot, P.; Brisse, H.; Philippe-Chomette, P.; Sarnacki, S.; Boccon-Gibod, L.; Rossignol, S.; Baumann, C.; et al. Malformations, genetic abnormalities, and Wilms tumor. Pediatr. Blood Cancer 2014, 61, 140–144. [Google Scholar] [CrossRef]
- Bliek, J.; Maas, S.; Alders, M.; Merks, J.H.; Mannens, M. Epigenotype, phenotype, and tumors in patients with isolated hemihyperplasia. J. Pediatr. 2008, 153, 95–100. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardinal Features (2 Points Each) | Suggestive Features (1 Point Each) |
|---|---|
| Macroglossia | Macrosomia (height/Birth Weight > +2SD) |
| Exomphalos | Facial naevus simplex |
| Lateralized overgrowth | Polyhydramnios/Placentomegaly |
| Multifocal/bilateral Wilms tumor or Nephroblastomatosis | Ear creases/pits |
| Hyperinsulinism | Transient hypoglycemia/hyperinsulinism |
| Pathology findings: | Typical BWSp tumors (neuroblastoma, rhabdomyosarcoma, unilateral WT, hepatoblastoma, adrenocortical carcinoma, phaeochromocytoma) |
| Adrenal cortex cytomegaly | Nephromegaly/Hepatomegaly |
| Placental mesenchymal dysplasia | Umbilical hernia/Diastasis recti |
| Pancreatic adenomatosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mussa, A.; Carli, D.; Cardaropoli, S.; Ferrero, G.B.; Resta, N. Lateralized and Segmental Overgrowth in Children. Cancers 2021, 13, 6166. https://doi.org/10.3390/cancers13246166
Mussa A, Carli D, Cardaropoli S, Ferrero GB, Resta N. Lateralized and Segmental Overgrowth in Children. Cancers. 2021; 13(24):6166. https://doi.org/10.3390/cancers13246166
Chicago/Turabian StyleMussa, Alessandro, Diana Carli, Simona Cardaropoli, Giovanni Battista Ferrero, and Nicoletta Resta. 2021. "Lateralized and Segmental Overgrowth in Children" Cancers 13, no. 24: 6166. https://doi.org/10.3390/cancers13246166
APA StyleMussa, A., Carli, D., Cardaropoli, S., Ferrero, G. B., & Resta, N. (2021). Lateralized and Segmental Overgrowth in Children. Cancers, 13(24), 6166. https://doi.org/10.3390/cancers13246166