
Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Cell Culture
2.3. Immunocytochemistry (ICC)
2.4. Confocal Imaging
2.5. DNA Isolation
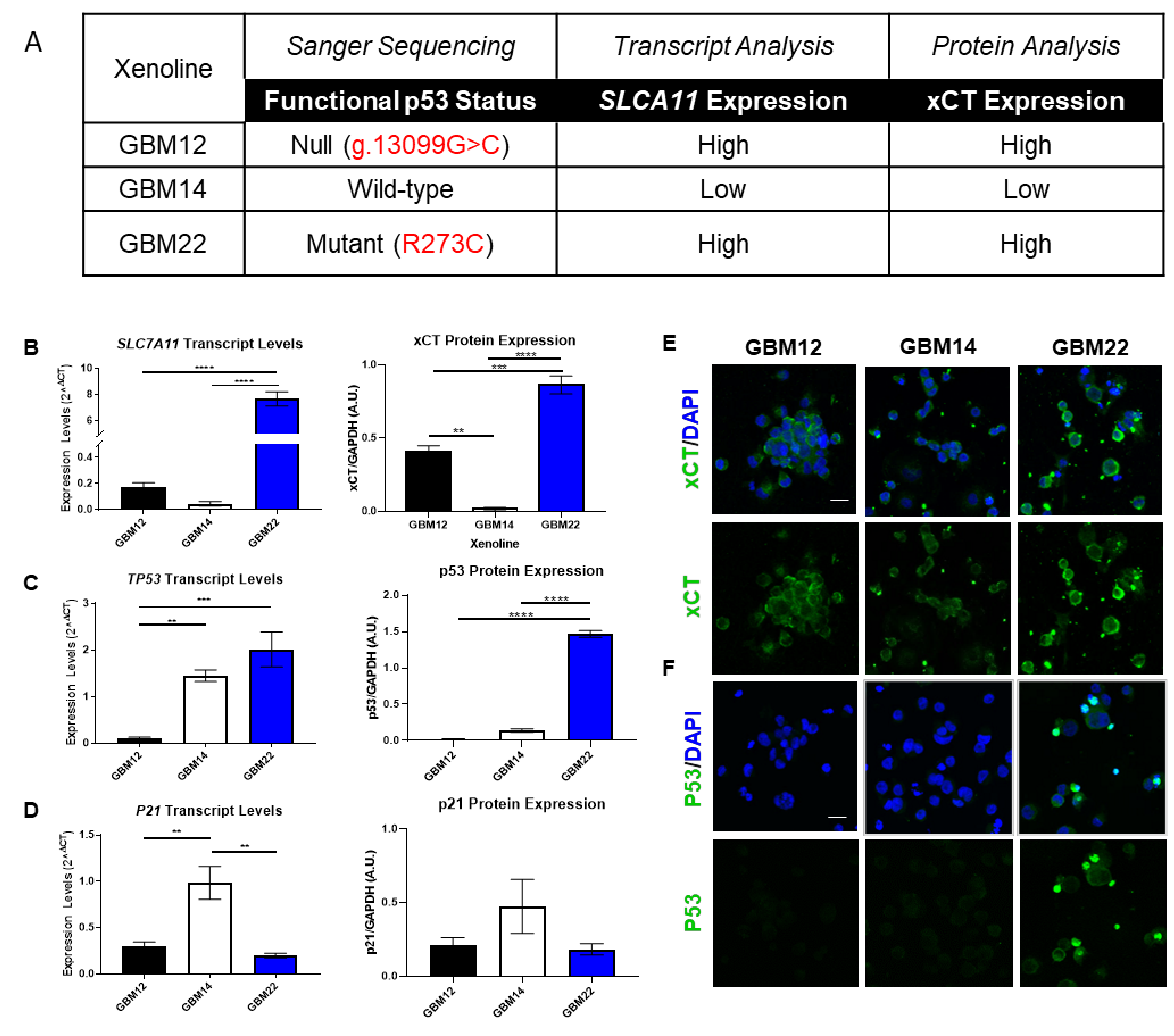
2.6. TP53 PCR and Sanger Sequencing
2.7. Protein Quantification and Western Blot
2.8. RT-qPCR
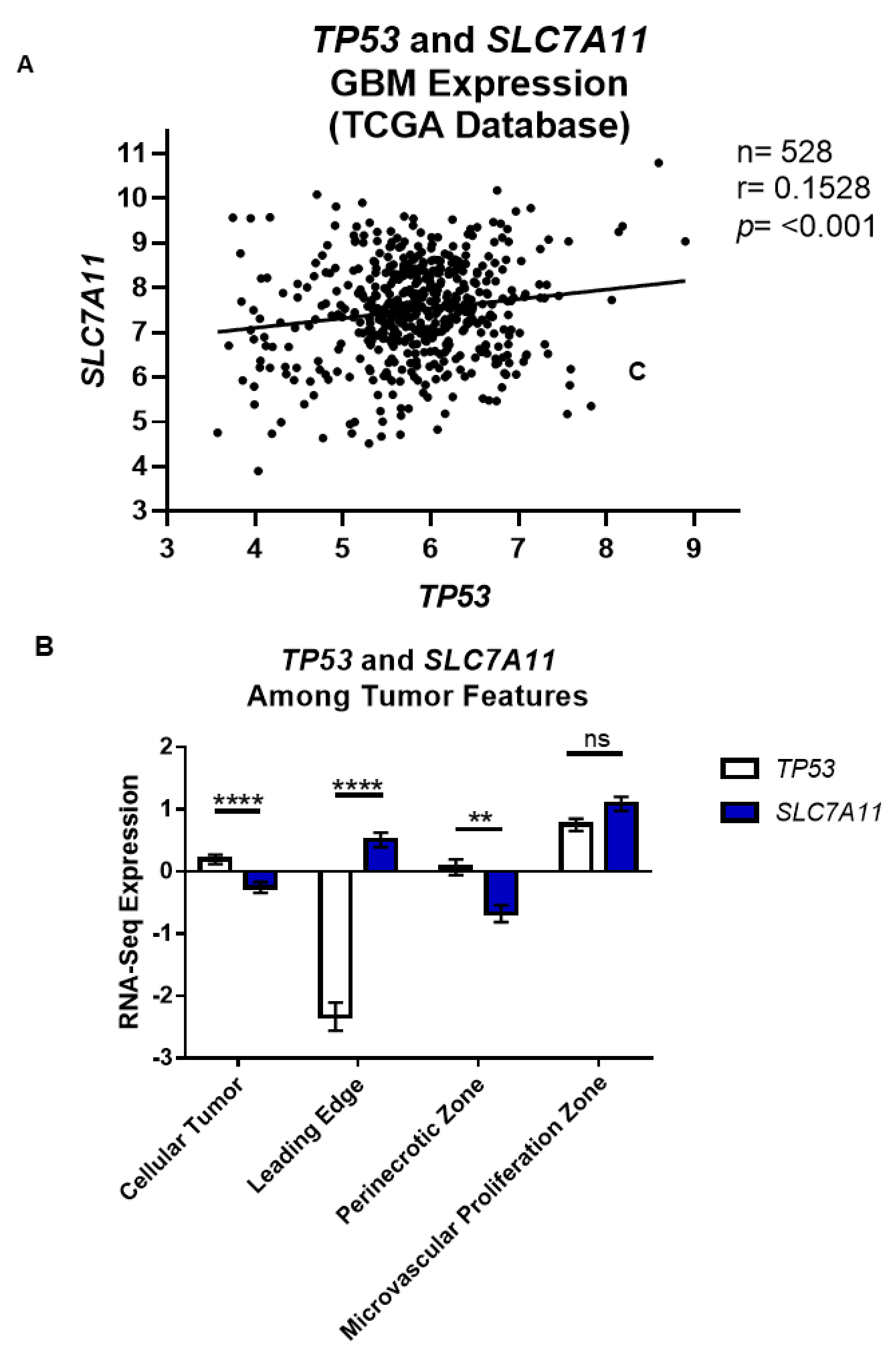
2.9. Database Mining
2.10. Chromatin Immunoprecipitation (ChIP) and PCR
2.11. siRNA Transfection
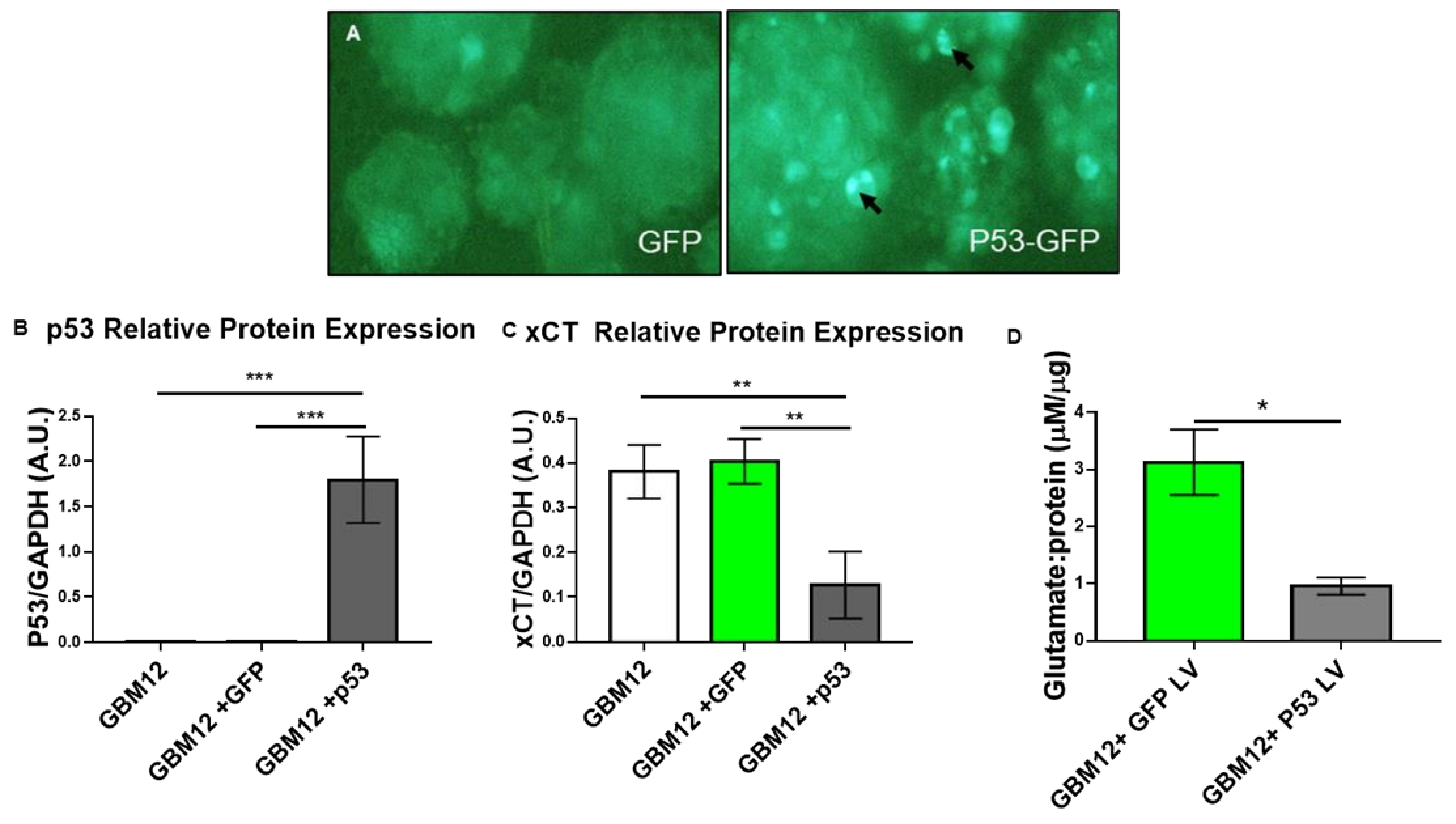
2.12. Lentiviral Transduction
2.13. Glutamate Release
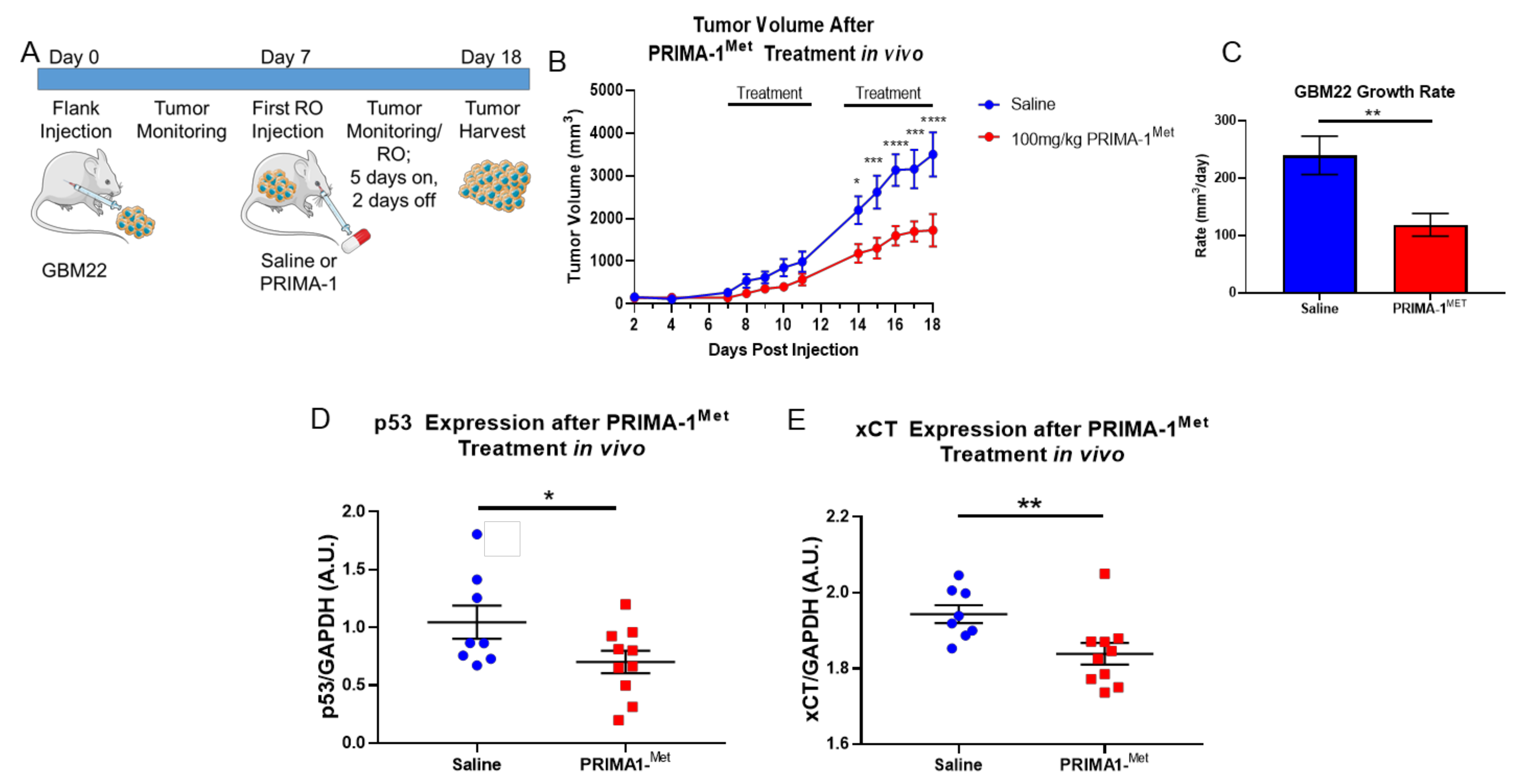
2.14. PRIMA-1Met Flank Tumor Experiments
2.15. PRIMA-1Met Intracranial Tumor Experiments
2.16. Statistics
3. Results
3.1. p53 Functional Status Is Relative to SLC7A11 Expression in GBM
3.2. Wild-Type p53 Is a Transcriptional Repressor of SLC7A11 Expression in Glioma
3.3. p53 Is a Molecular Toggle for SXC Glioma Biology
3.4. PRIMA-1Met Mitigates GBM Tumor Burden and xCT Expression In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. Jama 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.O.; Stupp, R. Epilepsy in brain tumor patients. Curr. Opin. Neurol. 2010, 23, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 289ra286. [Google Scholar] [CrossRef] [Green Version]
- Robert, S.M.; Sontheimer, H. Glutamate transporters in the biology of malignant gliomas. Cell Mol. Life Sci. 2014, 71, 1839–1854. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Sontheimer, H.; Bridges, R.J. Sulfasalazine for brain cancer fits. Expert Opin. Investig. Drugs 2012, 21, 575–578. [Google Scholar] [CrossRef]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of Cystine Uptake Disrupts the Growth of Primary Brain Tumors. J. Neurosci. 2005, 25, 7101. [Google Scholar] [CrossRef]
- Klotz, U. Clinical pharmacokinetics of sulphasalazine, its metabolites and other prodrugs of 5-aminosalicylic acid. Clin. Pharm. 1985, 10, 285–302. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Roshandel, A.K.; Busch, C.M.; Mullekom, J.V.; Cuoco, J.A.; Rogers, C.M.; Apfel, L.S.; Marvin, E.A.; Sontheimer, H.W.; Umans, R.A. The predictive capability of immunohistochemistry and DNA sequencing for determining TP53 functional mutation status: A comparative study of 41 glioblastoma patients. Oncotarget 2019, 10, 6204. [Google Scholar] [CrossRef] [Green Version]
- GlioVis: Data Visualization Tools for Brain Tumor Datasets. Available online: http://gliovis.bioinfo.cnio.es/ (accessed on 1 August 2019).
- IVY GAP: IVY Glioblastoma Atlas Project. Available online: https://glioblastoma.alleninstitute.org/ (accessed on 1 August 2019).
- Read, K.A.; Powell, M.D.; Baker, C.E.; Sreekumar, B.K.; Ringel-Scaia, V.M.; Bachus, H.; Martin, R.E.; Cooley, I.D.; Allen, I.C.; Ballesteros-Tato, A.; et al. Integrated STAT3 and Ikaros Zinc Finger transcription factor activities regulate Bcl-6 expression in CD4+ T helper cells. J. Immunol. 2017, 199, 2377–2387. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumaran, R.; Tan, K.H.; Miranda, P.J.; Haupt, S.; Haupt, Y. Regulation of Mutant p53 Protein Expression. Front Oncol. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Ye, Z.C.; Sontheimer, H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999, 59, 4383–4391. [Google Scholar]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, N.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y.; et al. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(-). Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawacka-Pankau, J.; Selivanova, G. Pharmacological reactivation of p53 as a strategy to treat cancer. J. Intern. Med. 2015, 277, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased Expression of System xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol. Cancer Res. 2016, 14, 1229–1242. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front Oncol. 2017, 7, 273. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Kharkar, P.S.; Gandhi, N.S.; Kaur, E.; Dutt, S.; Nandave, M. Novel analogs of sulfasalazine as system xc− antiporter inhibitors: Insights from the molecular modeling studies. Drug Dev. Res. 2019, 80, 758–777. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.-F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Ranjan, A.; Iwakuma, T. Emerging Non-Canonical Functions and Regulation of p53. Int. J. Mol. Sci. 2018, 19, 1015. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, M.; Vousden, K.H. Regulation of p53 stability. Oncogene 1999, 18, 7637–7643. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Banjac Canak, A.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [Green Version]
- Mlakar, V.; Jurkovic Mlakar, S.; Lesne, L.; Marino, D.; Rathi, K.S.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. PRIMA-1MET-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. J. Exp. Clin. Cancer Res. 2019, 38, 69. [Google Scholar] [CrossRef] [Green Version]
- Magri, J.; Gasparetto, A.; Conti, L.; Calautti, E.; Cossu, C.; Ruiu, R.; Barutello, G.; Cavallo, F. Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies. Cells 2021, 10, 108. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Smart Servier Medical Art. Available online: https://smart.servier.com (accessed on 1 August 2019).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umans, R.A.; Martin, J.; Harrigan, M.E.; Patel, D.C.; Chaunsali, L.; Roshandel, A.; Iyer, K.; Powell, M.D.; Oestreich, K.; Sontheimer, H. Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme. Cancers 2021, 13, 6169. https://doi.org/10.3390/cancers13246169
Umans RA, Martin J, Harrigan ME, Patel DC, Chaunsali L, Roshandel A, Iyer K, Powell MD, Oestreich K, Sontheimer H. Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme. Cancers. 2021; 13(24):6169. https://doi.org/10.3390/cancers13246169
Chicago/Turabian StyleUmans, Robyn A., Joelle Martin, Megan E. Harrigan, Dipan C. Patel, Lata Chaunsali, Aarash Roshandel, Kavya Iyer, Michael D. Powell, Ken Oestreich, and Harald Sontheimer. 2021. "Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme" Cancers 13, no. 24: 6169. https://doi.org/10.3390/cancers13246169