
Barriers to Immunotherapy in Ovarian Cancer: Metabolic, Genomic, and Immune Perturbations in the Tumour Microenvironment

 , , and
, , and 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Immunoregulatory Pathways within the TME
2.1. The Adaptive Immune Response
2.1.1. CTLA-4
2.1.2. PD-1
2.1.3. Predictors of ICB Response
2.1.4. ICB Combination Therapies
2.1.5. Other Adaptive Immunotherapeutic Strategies
2.2. The Innate Immune Response
2.2.1. Dendritic Cells
2.2.2. Natural Killer Cells
2.2.3. Macrophages
2.2.4. Myeloid-Derived Suppressor Cells
3. Tumour Cell Intrinsic Pathways and the Immunosuppressive TME
3.1. TP53
3.2. KRAS and PTEN
4. Metabolic Profile of the TME
4.1. Glucose and Lactate
4.2. Lipids
4.3. Hypoxia Is a Key Modulator of the TME
4.4. Amino Acids Metabolism and Immune Suppression in the TME
4.4.1. Glutamine
4.4.2. Arginine
4.4.3. Tryptophan
5. Concluding Remarks and the Future of Immunotherapy in OC
Author Contributions
Funding
Conflicts of Interest
References
- Ovarian Cancer Statistics. Cancer Research UK. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/ovarian-cancer (accessed on 26 August 2021).
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.A.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-year overall survival for patients with advanced non-small-cell lung cancer treated with pembrolizumab: Results from the phase i KEYNOTE-001 study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.A.; Elzey, B.D.; Hahn, N.M. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum. Vaccines Immunother. 2012, 8, 534–539. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Pakish, J.B.; Jazaeri, A.A. Immunotherapy in Gynecologic Cancers: Are We There Yet? Curr. Treat. Options Oncol. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Lin, J.-X.; Leonard, W.J. The Common Cytokine Receptor γ Chain Family of Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028449. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Kalekar, L.A.; Rosenblum, M.D. Regulatory T cells in inflammatory skin disease: From mice to humans. Int. Immunol. 2019, 31, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, N.; Tardiel-Cyril, D.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3 + Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Hermans, C.; Anz, D.; Engel, J.; Kirchner, T.; Endres, S.; Mayr, D. Analysis of FoxP3+ T-regulatory cells and CD8+T-Cells in ovarian carcinoma: Location and tumor infiltration patterns are key prognostic markers. PLoS ONE 2014, 9, e111757. [Google Scholar] [CrossRef] [Green Version]
- Hanaizi, Z.; van Zwieten-Boot, B.; Calvo, G.; Lopez, A.S.; van Dartel, M.; Camarero, J.; Abadie, E.; Pignatti, F. The European Medicines Agency review of ipilimumab (Yervoy) for the treatment of advanced (unresectable or metastatic) melanoma in adults who have received prior therapy: Summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur. J. Cancer 2012, 48, 237–242. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Phase II Study of Ipilimumab Monotherapy in Recurrent Platinum-Sensitive Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01611558 (accessed on 12 October 2021).
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Kirkwood, J.M.; Marshall, J.L.; Pritchard, C.C.; Azad, N.S.; Gulley, J.L. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J. Immunother. Cancer 2018, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Chang, M.; Chang, H.M.; Chang, F. Microsatellite Instability: A Predictive Biomarker for Cancer Immunotherapy. Appl. Immunohistochem. Mol. Morphol. AIMM 2018, 26, e15–e21. [Google Scholar] [CrossRef]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.-P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti–Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair–Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Kumar, A.; Sellers, T.A. Systematic review and meta-analysis of ovarian cancers: Estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin. Cancer Res. 2008, 14, 6847–6854. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Gordon, M.; Veneris, J.; Braiteh, F.; Balmanoukian, A.; Eder, J.P.; Oaknin, A.; Hamilton, E.; Wang, Y.; Sarkar, I.; et al. Safety, clinical activity and biomarker assessments of atezolizumab from a Phase I study in advanced/recurrent ovarian and uterine cancers. Gynecol. Oncol. 2019, 154, 314–322. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 154, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in patients with programmed death ligand 1–positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.F.; McDermott, D.F.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J., Jr.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized phase II trial of nivolumab versus nivolumab and ipilimumab for recurrent or persistent ovarian cancer: An NRG oncology study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca Clinical Trials A Phase Ib Study to Evaluate the Safety and Tolerability of Durvalumab and Tremelimumab in Combination with First-Line Chemotherapy in Patients with Advanced Solid Tumors. Available online: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=22730 (accessed on 28 September 2021).
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.R.; Thakkar, K.N.; Qian, J.; Kariolis, M.S.; Huang, W.; Nandagopal, S.; Yang, T.T.C.; Diep, A.N.; Cherf, G.M.; Xu, Y.; et al. Neutralization of PD-L2 is Essential for Overcoming Immune Checkpoint Blockade Resistance in Ovarian Cancer. Clin. Cancer Res. 2021, 27, 4435–4448. [Google Scholar] [CrossRef]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.M.; Marabelle, A.; Soria, J.-C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef] [Green Version]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Akbay, E.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Eldershaw, S.; Verma, K.; Croft, W.; Rai, T.; Kinsella, F.A.M.; Stephens, C.; Chen, L.; Nunnick, J.; Zuo, J.; Malladi, R.; et al. Lymphopenia-induced lymphoproliferation drives activation of naive T cells and expansion of regulatory populations. iScience 2021, 24, 102164. [Google Scholar] [CrossRef]
- Lutsiak, M.E.C.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.S.; Schlom, J.; Sabzevari, H. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.; Ray-Coquard, I.-L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): An open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 2021, 22, 1034–1046. [Google Scholar] [CrossRef]
- Monk, B.J.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 1275–1289. [Google Scholar] [CrossRef]
- Nishio, S.; Matsumoto, K.; Takehara, K.; Kawamura, N.; Hasegawa, K.; Takeshima, N.; Aoki, D.; Kamiura, S.; Arakawa, A.; Kondo, E.; et al. Pembrolizumab monotherapy in Japanese patients with advanced ovarian cancer: Subgroup analysis from the KEYNOTE-100. Cancer Sci. 2020, 111, 1324–1332. [Google Scholar] [CrossRef]
- Kim, C.; Wang, X.-D.; Yu, Y. Parp1 inhibitors trigger innate immunity via parp1 trapping-induced DNA damage response. eLife 2020, 9, 1–47. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 106. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2019, 30, v485–v486. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019, 79, 311. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Christopoulou, A.; Marmé, F.; Oaknin, A.; Lorusso, D.; Safra, T.; Lindahl, G.; Chudecka-Glaz, A.; Ciuleanu, T.; Collins, D.; Demirkiran, F.; et al. P116 ATHENA (GOG-3020/ENGOT-ov45; EudraCT 2017–004557–17; NCT03522246): A randomised, double-blind, placebo-controlled, phase 3 study of the poly(ADP-Ribose) polymerase (PARP) inhibitor rucaparib + the PD-1 inhibitor nivolumab following frontline platinum-based chemotherapy in ovarian cancer (OC). Int. J. Gynecol. Cancer 2019, 29, A130–A131. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Phase 1-2 Study of the Combination of Olaparib and Tremelimumab, in BRCA1 and BRCA2 Mutation Carriers with Recurrent Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02571725 (accessed on 28 September 2021).
- Moghaddam, S.M.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012, 31, 143–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartenbach, E.; Olson, T.; Goswitz, J.; Mohanraj, D.; Twiggs, L.; Carson, L.; Ramakrishnan, S. Vascular endothelial growth factor (VEGF) expression and survival in human epithelial ovarian carcinomas. Cancer Lett. 1997, 121, 169–175. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J.; et al. Efficacy and Safety of Pembrolizumab in Combination With Bevacizumab and Oral Metronomic Cyclophosphamide in the Treatment of Recurrent Ovarian Cancer: A Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 78–85. [Google Scholar] [CrossRef]
- Harter, P.; Bidziński, M.; Colombo, N.; Floquet, A.; Pérez, M.J.R.; Kim, J.-W.; Lheureux, S.; Marth, C.; Nyvang, G.-B.; Okamoto, A.; et al. DUO-O: A randomized phase III trial of durvalumab (durva) in combination with chemotherapy and bevacizumab (bev), followed by maintenance durva, bev and olaparib (olap), in newly diagnosed advanced ovarian cancer patients. J. Clin. Oncol. 2019, 37, TPS5598. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Retèl, V.P.; Steuten, L.M.G.; Foppen, M.H.G.; Mewes, J.C.; Lindenberg, M.A.; Haanen, J.B.A.G.; Van Harten, W.H. Early cost-effectiveness of tumor infiltrating lymphocytes (TIL) for second line treatment in advanced melanoma: A model-based economic evaluation. BMC Cancer 2018, 18, 895. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Ikarashi, H.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T. Prolonged Disease-Free Period in Patients with Advanced Epithelial Ovarian Cancer after Adoptive Transfer of Tumor-Infiltrating Lymphocytes. Clin. Cancer Res. 1995, 1, 501–507. [Google Scholar]
- Friese, C.; Harbst, K.; Borch, T.H.; Westergaard, M.C.W.; Pedersen, M.; Kverneland, A.; Jönsson, G.; Donia, M.; Svane, I.M.; Met, Ö. CTLA-4 blockade boosts the expansion of tumor-reactive CD8+ tumor-infiltrating lymphocytes in ovarian cancer. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kverneland, A.H.; Pedersen, M.; Wulff Westergaard, M.C.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Patterson, A.; Johnson, C.B.; Neitzke, D.J.; Mehrotra, S.; Denlinger, C.E.; Paulos, C.M.; Li, Z.; Cole, D.J.; Rubinstein, M.P. IL-2 and Beyond in Cancer Immunotherapy. J. Interf. Cytokine Res. 2018, 38, 45. [Google Scholar] [CrossRef]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: A pilot study. Oncoimmunology 2018, 7, e1502905. [Google Scholar] [CrossRef] [Green Version]
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignali, D.A. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin. Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef]
- Salas-Benito, D.; Conde, E.; Tamayo-Uria, I.; Mancheño, U.; Elizalde, E.; Garcia-Ros, D.; Aramendia, J.M.; Muruzabal, J.C.; Alcaide, J.; Guillen-Grima, F.; et al. The mutational load and a T-cell inflamed tumour phenotype identify ovarian cancer patients rendering tumour-reactive T cells from PD-1+ tumour-infiltrating lymphocytes. Br. J. Cancer 2021, 124, 1138–1149. [Google Scholar] [CrossRef]
- ClinicaTrials.gov. T-cell Therapy in Combination with Nivolumab, Relatlimab and Ipilimumab for Patients with Advanced Ovarian-, Fallopian Tube- and Primary Peritoneal Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04611126 (accessed on 28 September 2021).
- Osorio Ovarian Tumor Tissue Analysis (OTTA) Consortium; Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.; Chen, W.; Fogarty, Z.; Gentry-Maharaj, A.; Toloczko, A.; Hein, A.; et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef] [Green Version]
- Lopes, G.D.L.; Nahas, G.R. Chimeric antigen receptor T cells, a savior with a high price. Chin. Clin. Oncol. 2018, 7, 21. [Google Scholar] [CrossRef]
- Yen, M.J.; Hsu, C.-Y.; Mao, T.-L.; Wu, T.-C.; Roden, R.; Wang, T.-L.; Shih, L.-M. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin. Cancer Res. 2006, 12, 827–831. [Google Scholar] [CrossRef] [Green Version]
- Tanyi, J.L.; Haas, A.R.; Beatty, G.L.; Stashwick, C.J.; O’Hara, M.H.; Morgan, M.A.; Porter, D.L.; Melenhorst, J.J.; Plesa, G.; Lacey, S.F.; et al. Anti-mesothelin chimeric antigen receptor T cells in patients with epithelial ovarian cancer. J. Clin. Oncol. 2016, 34, 5511. [Google Scholar] [CrossRef]
- Figini, M.; Ferri, R.; Mezzanzanica, D.; Bagnoli, M.; Luison, E.; Miotti, S.; Canevari, S. Reversion of transformed phenotype in ovarian cancer cells by intracellular expression of anti folate receptor antibodies. Gene Ther. 2003, 10, 1018–1025. [Google Scholar] [CrossRef] [Green Version]
- Song, D.-G.; Ye, Q.; Carpenito, C.; Poussin, M.; Wang, L.P.; Ji, C.; Figini, M.; June, C.H.; Coukos, G.; Powell, D.J., Jr. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011, 71, 4617–4627. [Google Scholar] [CrossRef] [Green Version]
- Kandalaft, L.E.; Powell, D.J.; Coukos, G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J. Transl. Med. 2012, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.A.; Byrd, K.; Vreeland, T.J.; Clifton, G.T.; Jackson, D.O.; Hale, D.F.; Herbert, G.S.; Myers, J.W.; Greene, J.M.; Berry, J.S.; et al. Final analysis of a phase I/IIa trial of the folate-binding protein-derived E39 peptide vaccine to prevent recurrence in ovarian and endometrial cancer patients. Cancer Med. 2019, 8, 4678–4687. [Google Scholar] [CrossRef]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef]
- Harimoto, H.; Shimizu, M.; Nakagawa, Y.; Nakatsuka, K.; Wakabayashi, A.; Sakamoto, C.; Takahashi, H. Inactivation of tumor-specific CD8+CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunol. Cell Biol. 2013, 91, 545–555. [Google Scholar] [CrossRef]
- Conrad, C.; Gregorio, J.; Wang, Y.-H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.-J.; et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3+ T-regulatory cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [Green Version]
- Harada, Y.; Yonemitsu, Y. Recent developments in patented DC-based immunotherapy for various malignancies. Recent Pat. Regen. Med. 2011, 1, 72–87. [Google Scholar] [CrossRef]
- Luo, H.; Xu, X.; Ye, M.; Sheng, B.; Zhu, X. The prognostic value of HER2 in ovarian cancer: A meta-analysis of observational studies. PLoS ONE 2018, 13, e0191972. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.S.; Boyer, J.; Schullery, D.S.; Gimotty, P.A.; Gamerman, V.; Bender, J.; Levine, B.L.; Coukos, G.; Rubin, S.C.; Morgan, M.A.; et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol. Immunother. 2012, 61, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Block, M.S.; Dietz, A.B.; Gustafson, M.P.; Kalli, K.R.; Erskine, C.L.; Youssef, B.; Vijay, G.V.; Allred, J.B.; Pavelko, K.D.; Strausbauch, M.A.; et al. Th17-inducing autologous dendritic cell vaccination promotes antigen-specific cellular and humoral immunity in ovarian cancer patients. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Correale, P.; Cusi, M.G.; Del Vecchio, M.T.; Aquino, A.; Prete, S.; Tsang, K.Y.; Micheli, L.; Nencini, C.; La Placa, M.; Montagnani, F.; et al. Dendritic cell-mediated cross-presentation of antigens derived from colon carcinoma cells exposed to a highly cytotoxic multidrug regimen with gemcitabine, oxaliplatin, 5-fluorouracil, and leucovorin, elicits a powerful human antigen-specific CTL response with antitumor activity in vitro. J. Immunol. 2005, 175, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Cibula, D.; Rob, L.; Mallmann, P.; Knapp, P.; Klat, J.; Chovanec, J.; Minar, L.; Melichar, B.; Hein, A.; Kieszko, D.; et al. Dendritic cell-based immunotherapy (DCVAC/OvCa) combined with second-line chemotherapy in platinum-sensitive ovarian cancer (SOV02): A randomized, open-label, phase 2 trial. Gynecol. Oncol. 2021, 162, 652–660. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef] [Green Version]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [Green Version]
- Toubaji, A.; Hill, S.; Terabe, M.; Qian, J.; Floyd, T.; Simpson, R.M.; Berzofsky, J.A.; Khleif, S.N. The combination of GM-CSF and IL-2 as local adjuvant shows synergy in enhancing peptide vaccines and provides long term tumor protection. Vaccine 2007, 25, 5882–5891. [Google Scholar] [CrossRef]
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol. Immunother. 2012, 61, 373–384. [Google Scholar] [CrossRef] [Green Version]
- ClinicaTrials.gov. A Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial of DCVAC/OvCa Added to Standard of Care in Patients with Relapsed Platinum-sensitive Ovarian, Fallopian Tube, and Primary Peritoneal Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03905902 (accessed on 28 September 2021).
- Pallmer, K.; Oxenius, A. Recognition and Regulation of T Cells by NK Cells. Front. Immunol. 2016, 7, 251. [Google Scholar] [CrossRef] [Green Version]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology 2015, 4, e1001224. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Yao, Z.; Zhao, Z.; Xiao, H.; Xia, M.; Zhu, X.; Jiang, X.; Sun, C. Natural killer cells inhibit metastasis of ovarian carcinoma cells and show therapeutic effects in a murine model of ovarian cancer. Exp. Ther. Med. 2018, 16, 1071–1078. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Phase 3 Randomized, Double-Blind, Multicenter, Global Study of Monalizumab or Placebo in Combination with Cetuximab in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck Previously Treated with an Immune Checkpoint Inhibi. Available online: https://clinicaltrials.gov/ct2/show/NCT04590963 (accessed on 28 September 2021).
- Tinker, A.V.; Hirte, H.W.; Provencher, D.; Butler, M.; Ritter, H.; Tu, D.; Azim, H.A.; Paralejas, P.; Grenier, N.; Hahn, S.-A.; et al. Dose-Ranging and Cohort-Expansion Study of Monalizumab (IPH2201) in Patients with Advanced Gynecologic Malignancies: A Trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res. 2019, 25, 6052–6060. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Oaknin, A.; Sanchez-Simon, I.; Salgado, A.C.; Patel, S.P.; Oza, A.; Das, M.; Kourtesis, P.; Ascierto, M.L.; Diamond, J.R. 518 Phase 1B trial of monalizumab (NKG2A inhibitor) plus durvalumab: Safety and efficacy in patients with metastatic ovarian, cervical, and microsatellite-stable endometrial cancers. Int. J. Gynecol. Cancer 2020, 30, A86–A87. [Google Scholar] [CrossRef]
- Cooley, S.; He, F.; Bachanova, V.; Vercellotti, G.M.; DeFor, T.E.; Curtsinger, J.M.; Robertson, P.; Grzywacz, B.; Conlon, K.C.; Waldmann, T.A.; et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019, 3, 1970–1980. [Google Scholar] [CrossRef]
- Seo, H.; Jeon, I.; Kim, B.S.; Park, M.; Bae, E.A.; Song, B.; Koh, C.H.; Shin, K.S.; Kim, I.K.; Choi, K.; et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-Tumour immunity in MHC class I-deficient tumours. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Lin, M.; Luo, H.; Liang, S.; Chen, J.; Liu, A.; Niu, L.; Jiang, Y. Pembrolizumab plus allogeneic NK cells in advanced non-small cell lung cancer patients. J. Clin. Investig. 2020, 130, 2560–2569. [Google Scholar] [CrossRef]
- Hoogstad-van Evert, J.; Bekkers, R.; Ottevanger, N.; Schaap, N.; Hobo, W.; Jansen, J.H.; Massuger, L.; Dolstra, H. Intraperitoneal infusion of ex vivo-cultured allogeneic NK cells in recurrent ovarian carcinoma patients (a phase I study). Medicine 2019, 98, e14290. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Intraperitoneal FATE FT516 and Interleukin-2 (IL-2) with Intravenous Enoblituzumab in Recurrent Ovarian, Fallopian Tube, and Primary Peritoneal Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04630769 (accessed on 28 September 2021).
- Tonetti, C.R.; de Souza-Araújo, C.N.; Yoshida, A.; da Silva, R.F.; Alves, P.C.M.; Mazzola, T.N.; Derchain, S.; Fernandes, L.G.R.; Guimarães, F. Ovarian Cancer-Associated Ascites Have High Proportions of Cytokine-Responsive CD56bright NK Cells. Cells 2021, 10, 1702. [Google Scholar] [CrossRef]
- Wagner, J.A.; Rosario, M.; Romee, R.; Berrien-Elliott, M.; Schneider, S.E.; Leong, J.W.; Sullivan, R.P.; Jewell, B.A.; Becker-Hapak, M.; Schappe, T.; et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J. Clin. Investig. 2017, 127, 4042–4058. [Google Scholar] [CrossRef] [Green Version]
- Hoogstad-Van Evert, J.S.; Maas, R.J.; Van Der Meer, J.; Cany, J.; Van Der Steen, S.; Jansen, J.H.; Miller, J.S.; Bekkers, R.; Hobo, W.; Massuger, L.; et al. Peritoneal NK cells are responsive to IL-15 and percentages are correlated with outcome in advanced ovarian cancer patients. Oncotarget 2018, 9, 34810–34820. [Google Scholar] [CrossRef] [Green Version]
- Felices, M.; Chu, S.; Kodal, B.; Bendzick, L.; Ryan, C.; Lenvik, A.J.; Boylan, K.L.M.; Wong, H.C.; Skubitz, A.P.N.; Miller, J.S.; et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol. Oncol. 2017, 145, 453–461. [Google Scholar] [CrossRef]
- Geller, M.A.; Cooley, S.A.; Wallet, M.; Valamehr, B.; Teoh, D.G.K.; DeFor, T.E.; Felices, M.; Miller, J. APOLLO: A phase I study of adaptive memory natural killer (NK) cells in recurrent ovarian cancer. J. Clin. Oncol. 2020, 38, 6044. [Google Scholar] [CrossRef]
- Cooley, S.; Geller, M.; Cichocki, F.; Curtsinger, J.; McKenna, D.H.; Storgard, C.; Valamehr, B.; Miller, J.S. In Vivo Persistence and Function of Adaptive NK Cell Infusions (FATE-NK100) from CMV Seropositive Haploidentical Related Donors. Biol. Blood Marrow Transplant. 2019, 25, S338. [Google Scholar] [CrossRef]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [Green Version]
- McGilvray, R.W.; Eagle, R.A.; Rolland, P.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int. J. Cancer 2010, 127, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Mandai, M.; Hamanishi, J.; Matsumura, N.; Suzuki, A.; Yagi, H.; Yamaguchi, K.; Baba, T.; Fujii, S.; Konishi, I. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: High expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol. Immunother. 2009, 58, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Tay, J.C.K.; Wang, S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol. Ther.-Oncolytics 2020, 16, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y.; et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Clinical Study on the Safety and Efficacy of Anti-Mesothelin Car NK Cells with Epithelial Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03692637 (accessed on 28 September 2021).
- Baci, D.; Bosi, A.; Gallazzi, M.; Rizzi, M.; Noonan, D.M.; Poggi, A.; Bruno, A.; Mortara, L. The ovarian cancer tumor immune microenvironment (Time) as target for therapy: A focus on innate immunity cells as therapeutic effectors. Int. J. Mol. Sci. 2020, 21, 3125. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.-y.; Shi, H.-s.; Zeng, Y.-h.; Wang, X.-z.; Zhao, Y.-w.; Wei, Y.-q. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fingerle-Rowson, G.; Petrenko, O.; Metz, C.N.; Forsthuber, T.G.; Mitchell, R.; Huss, R.; Moll, U.; Muller, W.; Bucala, R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. USA 2003, 100, 9354–9359. [Google Scholar] [CrossRef] [Green Version]
- Krockenberger, M.; Dombrowski, Y.; Weidler, C.; Ossadnik, M.; Hönig, A.; Häusler, S.; Voigt, H.; Becker, J.C.; Leng, L.; Steinle, A.; et al. Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J. Immunol. 2008, 180, 7338–7348. [Google Scholar] [CrossRef]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages Induce Invasiveness of Epithelial Cancer Cells Via NF-κB and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Neyen, C.; Plüddemann, A.; Mukhopadhyay, S.; Maniati, E.; Bossard, M.; Gordon, S.; Hagemann, T. Macrophage Scavenger Receptor A Promotes Tumor Progression in Murine Models of Ovarian and Pancreatic Cancer. J. Immunol. 2013, 190, 3798–3805. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Soto-Pantoja, D.R.; Terabe, M.; Ghosh, A.; Ridnour, L.A.; DeGraff, W.G.; Wink, D.A.; Berzofsky, J.A.; Roberts, D.D. Cd47 in the tumor microenvironment limits cooperation between antitumor t-cell immunity and radiotherapy. Cancer Res. 2014, 74, 6771–6783. [Google Scholar] [CrossRef] [Green Version]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.M.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garciaa, K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2646–E2654. [Google Scholar] [CrossRef] [Green Version]
- Yanagita, T.; Murata, Y.; Tanaka, D.; Motegi, S.; Arai, E.; Daniwijaya, E.W.; Hazama, D.; Washio, K.; Saito, Y.; Kotani, T.; et al. Anti-SIRPα antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2017, 2, e89140. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Phase I/II Study Evaluating Safety and Clinical Efficacy of SHR2150 (TLR7 Agonist) in Combination with Chemotherapy Plus PD-1 or CD47 Antibody in Subjects with Unresectable/ Metastatic Solid Tumors. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04588324 (accessed on 28 September 2021).
- Eyvazi, S.; Kazemi, B.; Dastmalchi, S.; Bandehpour, M. Involvement of CD24 in Multiple Cancer Related Pathways Makes It an Interesting New Target for Cancer Therapy. Curr. Cancer Drug Targets 2018, 18, 328–336. [Google Scholar] [CrossRef]
- Davidson, B. CD24 is highly useful in differentiating high-grade serous carcinoma from benign and malignant mesothelial cells. Hum. Pathol. 2016, 58, 123–127. [Google Scholar] [CrossRef]
- Kristiansen, G.; Denkert, C.; Schlüns, K.; Dahl, E.; Pilarsky, C.; Hauptmann, S. CD24 is expressed in ovarian cancer and is a new independent prognostic marker of patient survival. Am. J. Pathol. 2002, 161, 1215–1221. [Google Scholar] [CrossRef] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Klapdor, R.; Wang, S.; Morgan, M.; Dörk, T.; Hacker, U.; Hillemanns, P.; Büning, H.; Schambach, A. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int. J. Mol. Sci. 2019, 20, 660. [Google Scholar] [CrossRef] [Green Version]
- Loges, S.; Schmidt, T.; Tjwa, M.; Van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [Green Version]
- Antony, J.; Tan, T.Z.; Kelly, Z.; Low, J.; Choolani, M.; Recchi, C.; Gabra, H.; Thiery, J.P.; Huang, R.Y.-J. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci. Signal. 2016, 9, ra97. [Google Scholar] [CrossRef]
- Antony, J.; Huang, R.Y.J. AXL-driven EMT state as a targetable conduit in cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Chen, X.S.; Li, L.Y.; Wu, H.Z.; Zeng, D.; Wang, X.L.; Zhang, Y.; Xiao, S.S.; Cheng, Y. Inhibition of AXL enhances chemosensitivity of human ovarian cancer cells to cisplatin via decreasing glycolysis. Acta Pharmacol. Sin. 2020, 42, 1180–1189. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Phase IB Study of Paclitaxel + Carboplatin with AVB-S6-500 in Women with Stage III or IV Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer Receiving Neoadjuvant Chemotherapy. Available online: https://clinicaltrials.gov/ct2/show/NCT03607955 (accessed on 28 September 2021).
- Paolino, M.; Penninger, J.M. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers 2016, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Spicer, J.; Helland, Å.; Carcereny, E.; Arriola, E.; Gomez, M.D.; Perez, J.M.T.; Thompson, J.; Strauss, J.; Granados, A.L.O.; Felip, E.; et al. 362A PhII study of bemcentinib, a first-in-class selective AXL kinase inhibitor, in combination with pembrolizumab in pts with previously-treated advanced NSCLC: Updated clinical & translational analysis. J. Immunother. Cancer 2020, 8, A387. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Randomized Phase I/II Study of AVB-S6-500 in Combination with Durvalumab (MEDI4736) in Patients with Platinum-Resistant, Recurrent Epithelial Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04019288 (accessed on 28 September 2021).
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Vicari, A.P.; Chiodoni, C.; Vaure, C.; Aït-Yahia, S.; Dercamp, C.; Matsos, F.; Reynard, O.; Taverne, C.; Merle, P.; Colombo, M.P.; et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J. Exp. Med. 2002, 196, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Sanaei, M.; Taheri, F.; Heshmati, M.; Bashash, D.; Nazmabadi, R.; Mohammad-Alibeigi, F.; Nahid-Samiei, M.; Shirzad, H.; Bagheri, N. Comparing the frequency of CD33 + pSTAT3 + myeloid-derived suppressor cells and IL-17 + lymphocytes in patients with prostate cancer and benign prostatic hyperplasia. Cell Biol. Int. 2021, 45, 2086–2095. [Google Scholar] [CrossRef]
- Brusa, D.; Simone, M.; Gontero, P.; Spadi, R.; Racca, P.; Micari, J.; Degiuli, M.; Carletto, S.; Tizzani, A.; Matera, L. Circulating immunosuppressive cells of prostate cancer patients before and after radical prostatectomy: Profile comparison. Int. J. Urol. 2013, 20, 971–978. [Google Scholar] [CrossRef]
- Coosemans, A.; Baert, T.; Ceusters, J.; Busschaert, P.; Landolfo, C.; Verschuere, T.; Van Rompuy, A.S.; Vanderstichele, A.; Froyman, W.; Neven, P.; et al. Myeloid-derived suppressor cells at diagnosis may discriminate between benign and malignant ovarian tumors. Int. J. Gynecol. Cancer 2019, 29, 1381–1388. [Google Scholar] [CrossRef]
- Baert, T.; Vankerckhoven, A.; Riva, M.; Van Hoylandt, A.; Thirion, G.; Holger, G.; Mathivet, T.; Vergote, I.; Coosemans, A. Myeloid derived suppressor cells: Key drivers of immunosuppression in ovarian cancer. Front. Immunol. 2019, 10, 1273. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Yang, L.; Xue, J.; Hu, G. Blockade of CCL2 enhances immunotherapeutic effect of anti-PD1 in lung cancer. J. Bone Oncol. 2018, 11, 27–32. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Phase 1, Open-Label, Dose-Escalation with Expansion Study of SX-682 in Subjects with Metastatic Melanoma Concurrently Treated with Pembrolizumab. Available online: https://clinicaltrials.gov/ct2/show/NCT03161431 (accessed on 28 September 2021).
- ClinicalTrials.gov. A Phase IIb Pilot Study to Assess the Efficacy, Safety, and Pharmacodynamics Effects of Pembrolizumab and BL-8040 in Patients with Metastatic Pancreatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02907099 (accessed on 28 September 2021).
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Phase 1 Study of RGX-104, a Small Molecule LXR Agonist, as a Single Agent and as Combination Therapy in Patients with Advanced Solid Malignancies and Lymphoma with an Expansion in Select Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT02922764 (accessed on 28 September 2021).
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kalamanathan, S.; Bates, V.; Lord, R.; Green, J.A. The mutational profile of sporadic epithelial ovarian carcinoma. Anticancer Res. 2011, 31, 2661–2668. [Google Scholar]
- Ramalingam, P. Morphologic, Immunophenotypic, and Molecular Features of Epithelial Ovarian Cancer. Oncology 2016, 30, 166–176. [Google Scholar]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, M.T.; Leone, E.; Amodio, N.; Foresta, U.; Lionetti, M.; Pitari, M.R.; Cantafio, M.E.G.; Gullà, A.; Conforti, F.; Morelli, E.; et al. Synthetic miR-34a mimics as a novel therapeutic agent for Multiple Myeloma: In vitro and in vivo evidence. Clin. Cancer Res. 2012, 18, 6260. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Leffers, N.; Lambeck, A.J.A.; Gooden, M.J.M.; Hoogeboom, B.N.; Wolf, R.; Hamming, I.E.; Hepkema, B.G.; Willemse, P.H.B.; Molmans, B.H.W.; Hollema, H.; et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int. J. Cancer 2009, 125, 2104–2113. [Google Scholar] [CrossRef]
- Leffers, N.; Vermeij, R.; Hoogeboom, B.N.; Schulze, U.R.; Wolf, R.; Hamming, I.E.; Van Der Zee, A.G.; Melief, K.J.; Van Der Burg, S.H.; Daemen, T.; et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int. J. Cancer 2012, 130, 105–112. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 1–12. [Google Scholar] [CrossRef]
- Hardwick, N.R.; Frankel, P.; Ruel, C.; Kilpatrick, J.; Tsai, W.; Kos, F.; Kaltcheva, T.; Leong, L.; Morgan, R.; Chung, V.; et al. p53-Reactive T Cells Are Associated with Clinical Benefit in Patients with Platinum-Resistant Epithelial Ovarian Cancer After Treatment with a p53 Vaccine and Gemcitabine Chemotherapy. Clin. Cancer Res. 2018, 24, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Vermeij, R.; Leffers, N.; Hoogeboom, B.N.; Hamming, I.L.E.; Wolf, R.; Reyners, A.K.L.; Molmans, B.H.W.; Hollema, H.; Bart, J.; Drijfhout, J.W.; et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int. J. Cancer 2012, 131, E670–E680. [Google Scholar] [CrossRef]
- Hardwick, N.R.; Carroll, M.; Kaltcheva, T.; Qian, D.; Lim, D.; Leong, L.; Chu, P.; Kim, J.; Chao, J.; Fakih, M.; et al. p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin. Cancer Res. 2014, 20, 4459–4470. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.; Kos, F.J.; Hardwick, N.; Yuan, Y.; Chao, J.; Li, D.; Waisman, J.; Li, M.; Zurcher, K.; Frankel, P.; et al. Evaluation of safety and efficacy of p53MVA vaccine combined with pembrolizumab in patients with advanced solid cancers. Clin. Transl. Oncol. 2019, 21, 363–372. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. P53MVA and Pembrolizumab in Treating Patients With Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03113487 (accessed on 28 September 2021).
- Maheswaran, S.; Park, S.; Bernard, A.; Morris, J.F.; Rauscher, F.J.; Hill, D.E.; Haber, D.A. Physical and functional interaction between WT1 and p53 proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 5100–5104. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.H.; Deddens, J.A.; Mueller, G.; Lewis, T.G.; Dooley, M.K.; Robillard, M.C.; Frydl, M.; Duvall, L.; Pemberton, J.O.; Douglass, L.E. Transcription factors wt1 and p53 combined: A prognostic biomarker in ovarian cancer. Br. J. Cancer 2018, 119, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, T.; Ueda, Y.; Morimoto, A.; Enomoto, T.; Nishida, S.; Shirakata, T.; Oka, Y.; Tsuboi, A.; Oji, Y.; Hosen, N.; et al. WT1 peptide immunotherapy for gynecologic malignancies resistant to conventional therapies: A phase II trial. J. Cancer Res. Clin. Oncol. 2013, 139, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Kyo, S.; Myojo, S.; Dohi, S.; Ishizaki, J.; Miyamoto, K.I.; Morita, S.; Sakamoto, J.I.; Enomoto, T.; Kimura, T.; et al. Wilms’ tumor 1 (WTl) peptide immunotherapy for gynecological malignancy. Anticancer Res. 2009, 29, 4779–4784. [Google Scholar] [PubMed]
- ClinicalTrials.gov. A Phase I Study of Concomitant WT1 Analog Peptide Vaccine or NY-ESO-1 Overlapping Peptides Vaccine in Combination with Nivolumab in Patients with Recurrent Ovarian Cancer Who Are in Second or Greater Remission. Available online: https://clinicaltrials.gov/ct2/show/NCT02737787 (accessed on 28 September 2021).
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 206–219. [Google Scholar] [CrossRef]
- Ji, L.; Xu, J.; Liu, J.; Amjad, A.; Zhang, K.; Liu, Q.; Zhou, L.; Xiao, J.; Li, X. Mutant p53 promotes tumor cell malignancy by both positive and negative regulation of the transforming growth factor β (TGF-β) pathway. J. Biol. Chem. 2015, 290, 11729–11740. [Google Scholar] [CrossRef] [Green Version]
- Levy, L.; Hill, C.S. Smad4 Dependency Defines Two Classes of Transforming Growth Factor β (TGF-β) Target Genes and Distinguishes TGF-β-Induced Epithelial-Mesenchymal Transition from Its Antiproliferative and Migratory Responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Kao, J.Y.; Gong, Y.; Chen, C.-M.; Zheng, Q.-D.; Chen, J.-J. Tumor-Derived TGF-β Reduces the Efficacy of Dendritic Cell/Tumor Fusion Vaccine. J. Immunol. 2003, 170, 3806–3811. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef]
- Oh, J.; Barve, M.; Senzer, N.; Aaron, P.; Manning, L.; Wallraven, G.; Bognar, E.; Stanbery, L.; Horvath, S.; Manley, M.; et al. Long-term follow-up of Phase 2A trial results involving advanced ovarian cancer patients treated with Vigil® in frontline maintenance. Gynecol. Oncol. Rep. 2020, 34, 100648. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Monk, B.J.; Walter, A.; Herzog, T.J.; Galanis, E.; Manning, L.; Bognar, E.; Wallraven, G.; Stanbery, L.; Aaron, P.; et al. Gemogenovatucel-T (Vigil) immunotherapy demonstrates clinical benefit in homologous recombination proficient (HRP) ovarian cancer. Gynecol. Oncol. 2021, 161, 676–680. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Grosen, E.A.; Ghamande, S.A.; Chan, J.K.; Barve, M.A.; Oh, J.; Tewari, D.; Morris, P.C.; Stevens, E.E.; Bottsford-Miller, J.N.; et al. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol. 2020, 21, 1661–1672. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Stevens, E.E.; Bottsford-Miller, J.N.; Ghamande, S.A.; Aaron, P.; Wallraven, G.; Bognar, E.; Manley, M.; Horvath, S.; Manning, L.; et al. A phase I combination study of vigil and atezolizumab in recurrent/refractory advanced-stage ovarian cancer: Efficacy assessment in BRCA1/2-wt patients. J. Clin. Oncol. 2020, 38, 3002. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 trans-signaling promotes KRAS-driven lung carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras-mutant lung cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.A.; de Carné Trécesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Phase 1b/2, Protocol Evaluating the Safety, Tolerability, Pharmacokinetics, and Efficacy of Sotorasib Monotherapy and in Combination with Other Anti-Cancer Therapies in Subjects with Advanced Solid Tumors with KRAS p.G12C Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT04185883 (accessed on 28 September 2021).
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.-J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes. Chromosomes Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Kinross, K.M.; Montgomery, K.G.; Kleinschmidt, M.; Waring, P.; Ivetac, I.; Tikoo, A.; Saad, M.; Hare, L.; Roh, V.; Mantamadiotis, T.; et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Investig. 2012, 122, 553–557. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Hofmann, J.; Lu, Y.; Mills, G.B.; Jaffe, R.B. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002, 62, 1087–1092. [Google Scholar]
- Hu, L.; Zaloudek, C.; Mills, G.B.; Gray, J.; Jaffe, R.B. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin. Cancer Res. 2000, 6, 880–886. [Google Scholar]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Behbakht, K.; Sill, M.W.; Darcy, K.M.; Rubin, S.C.; Mannel, R.S.; Waggoner, S.; Schilder, R.J.; Cai, K.Q.; Godwin, A.K.; Alpaugh, R.K. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: A Gynecologic Oncology Group study. Gynecol. Oncol. 2011, 123, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Wheler, J.; Mutch, D.; Lager, J.; Castell, C.; Liu, L.; Jiang, J.; Traynor, A.M. Phase I Dose-Escalation Study of Pilaralisib (SAR245408, XL147) in Combination with Paclitaxel and Carboplatin in Patients with Solid Tumors. Oncologist 2017, 22, 377. [Google Scholar] [CrossRef]
- Blagden, S.P.; Hamilton, A.L.; Mileshkin, L.; Wong, S.; Michael, A.; Hall, M.; Goh, J.C.; Lisyanskaya, A.S.; DeSilvio, M.; Frangou, E.; et al. Phase IB Dose Escalation and Expansion Study of AKT Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-resistant Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1472–1478. [Google Scholar] [CrossRef] [Green Version]
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib keynote-012 study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.-C.; Ayers, G.; Abramson, V.; Mayer, I.; Richmond, A. Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Phase I/II Biomarker Driven Combination Trial of Copanlisib and Immune Checkpoint Inhibitors in Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04317105 (accessed on 28 September 2021).
- Morschhauser, F.; Machiels, J.-P.; Salles, G.; Rottey, S.; Rule, S.A.J.; Cunningham, D.; Peyrade, F.; Fruchart, C.; Arkenau, H.-T.; Genvresse, I.; et al. On-Target Pharmacodynamic Activity of the PI3K Inhibitor Copanlisib in Paired Biopsies from Patients with Malignant Lymphoma and Advanced Solid Tumors. Mol. Cancer Ther. 2020, 19, 468–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Anjomani-Virmouni, S.; Koundouros, N.; Dimitriadi, M.; Choo-Wing, R.; Valle, A.; Zheng, Y.; Chiu, Y.-H.; Agnihotri, S.; Zadeh, G.; et al. PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol. Cell 2017, 65, 999–1013.e7. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Reports 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Najumudeen, A.K.; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; Race, A.M.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Xu, L.; Fidler, I.J. Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res. 2000, 60, 4610–4616. [Google Scholar]
- Shi, Q.; Le, X.; Wang, B.; Xiong, Q.; Abbruzzese, J.L.; Xie, K. Regulation of interleukin-8 expression by cellular pH in human pancreatic adenocarcinoma cells. J. Interf. Cytokine Res. 2000, 20, 1023–1028. [Google Scholar] [CrossRef]
- Xiang, J.; Zhou, L.; Zhuang, Y.; Zhang, J.; Sun, Y.; Li, S.; Zhang, Z.; Zhang, G.; He, Y. Lactate dehydrogenase is correlated with clinical stage and grade and is downregulated by si-SATB1 in ovarian cancer. Oncol. Rep. 2018, 40, 2788–2797. [Google Scholar] [CrossRef]
- Ikeda, A.; Yamaguchi, K.; Abiko, K.; Takakura, K.; Konishi, I.; Yamaguchi, K.; Yamaguchi, K.; Yamakage, H.; Satoh-Asahara, N. Serum lactate dehydrogenase is a possible predictor of platinum resistance in ovarian cancer. Obstet. Gynecol. Sci. 2020, 63, 709–718. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic Cell Reprogramming by Endogenously Produced Lactic Acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Yan, X.-Y.; Yu, W.-W.; Liang, X.-Q.; Du, X.-Y.; Liu, Z.-C.; Long, J.-P.; Zhao, G.-H.; Liu, H.-B. Lactic acid in macrophage polarization: The significant role in inflammation and cancer. Int. Rev. Immunol. 2021, 1–15. [Google Scholar] [CrossRef]
- Bosticardo, M.; Ariotti, S.; Losana, G.; Bernabei, P.; Forni, G.; Novelli, F. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur. J. Immunol. 2001, 31, 2829–2838. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; Van Der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. XPosttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct pd-1 + cd8 + t cell pool with predictive potential in non-small-cell lung cancer treated with pd-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e4. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [Green Version]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Gotlieb, W.H.; Saumet, J.; Beauchamp, M.-C.; Gu, J.; Lau, S.; Pollak, M.N.; Bruchim, I. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol. Oncol. 2008, 110, 246–250. [Google Scholar] [CrossRef]
- Rattan, R.; Graham, R.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 2011, 13, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-B.; Lei, K.-J.; Liu, J.-P.; Jia, Y.-M. Continuous use of metformin can improve survival in type 2 diabetic patients with ovarian cancer: A retrospective study. Medicine 2017, 96, e7605. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Curry, J.M.; Johnson, J.; Mollaee, M.; Tassone, P.; Amin, D.; Knops, A.; Whitaker-Menezes, D.; Mahoney, M.G.; South, A.; Rodeck, U.; et al. Metformin clinical trial in HPV+ and HPV-head and neck squamous cell carcinoma: Impact on cancer cell apoptosis and immune infiltrate. Front. Oncol. 2018, 8, 436. [Google Scholar] [CrossRef]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Mu, Q.; Jiang, M.; Zhang, Y.; Wu, F.; Li, H.; Zhang, W.; Wang, F.; Liu, J.; Li, L.; Wang, D.; et al. Metformin inhibits proliferation and cytotoxicity and induces apoptosis via AMPK pathway in CD19-chimeric antigen receptor-modified T cells. OncoTargets Ther. 2018, 11, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B. CD73: A novel target for cancer immunotherapy. Cancer Res. 2010, 70, 6407–6411. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Parallel Proof of Concept Phase 2 Study of Nivolumab and Metformin Combination Treatment in Advanced Non-Small Cell Lung Cancer with and without Prior Treatment with PD-1/PD-L1 Inhibitors. Available online: https://clinicaltrials.gov/ct2/show/NCT03048500 (accessed on 28 September 2021).
- ClinicalTrials.gov. Phase II Trial of Nivolumab and Metformin in Patients with Treatment Refractory MSS Metastatic Colorectal Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03800602 (accessed on 28 September 2021).
- ClinicalTrials.gov. A Phase 2 Feasibility Study Combining Pembrolizumab and Metformin to Harness the Natural Killer Cytotoxic Response in Metastatic Head and Neck Cancer Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT04414540 (accessed on 28 September 2021).
- ClinicalTrials.gov. Window of Opportunity for Durvalumab (MEDI4736) Plus Metformin Trial of in Squamous Cell Carcinoma of the Head and Neck. Available online: https://clinicaltrials.gov/ct2/show/NCT03618654 (accessed on 28 September 2021).
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Xu, B.; Zhu, N.; Ren, B. PGC-1α activator–induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori J. 2020, 106, 55–63. [Google Scholar] [CrossRef]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8 + T cell proliferation. Cell. Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef]
- Azpilikueta, A.; Agorreta, J.; Labiano, S.; Perez-Gracia, J.L.; Sánchez-Paulete, A.R.; Aznar, M.A.; Ajona, D.; Gil-Bazo, I.; Larrayoz, M.; Teijeira, A.; et al. Successful Immunotherapy against a Transplantable Mouse Squamous Lung Carcinoma with Anti-PD-1 and Anti-CD137 Monoclonal Antibodies. J. Thorac. Oncol. 2016, 11, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Chacon, J.A.; Li, Y.; Wu, R.C.; Bernatchez, C.; Wang, Y.; Weber, J.S.; Hwu, P.; Radvanyi, L.G. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J. Immunother. 2011, 34, 236–250. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Phase I/Ib Study of Adoptive Cellular Therapy Using Autologous IL-21-Primed CD8+ Tumor Antigen-Specific T Cells in Combination with Utomilumab (PF-05082566) in Patients with Platinum Resistant Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03318900 (accessed on 28 September 2021).
- ClinicalTrials.gov. A Study of Avelumab in Combination with Other Cancer Immunotherapies in Advanced Malignancies (JAVELIN Medley). Available online: https://clinicaltrials.gov/ct2/show/NCT02554812 (accessed on 28 September 2021).
- Innocenzi, D.; Alò, P.L.; Balzani, A.; Sebastiani, V.; Silipo, V.; La Torre, G.; Ricciardi, G.; Bosman, C.; Calvieri, S. Fatty acid synthase expression in melanoma. J. Cutan. Pathol. 2003, 30, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Kuhajda, F.P.; Jenner, K.; Wood, F.D.; Hennigar, R.A.; Jacobs, L.B.; Dick, J.D.; Pasternack, G.R. Fatty acid synthesis: A potential selective target for antineoplastic therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 6379–6383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Gansler, T.S.; Hardman, W.; Hunt, D.A.; Schaffel, S.; Hennigar, R.A. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum. Pathol. 1997, 28, 686–692. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef]
- Pampalakis, G.; Politi, A.-L.; Papanastasiou, A.; Sotiropoulou, G. Distinct cholesterogenic and lipidogenic gene expression patterns in ovarian cancer—A new pool of biomarkers. Genes Cancer 2015, 6, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Ayyagari, V.N.; Wang, X.; Diaz-Sylvester, P.L.; Groesch, K.; Brard, L. Assessment of acyl-CoA cholesterol acyltransferase (ACAT-1) role in ovarian cancer progression-An in vitro study. PLoS ONE 2020, 15, e0228024. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Angela, M.; Endo, Y.; Asou, H.K.; Yamamoto, T.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARγ directs early activation of T cells. Nat. Commun. 2016, 7, 13683. [Google Scholar] [CrossRef]
- Beziaud, L.; Mansi, L.; Ravel, P.; Marie-Joseph, E.L.; Laheurte, C.; Rangan, L.; Bonnefoy, F.; Pallandre, J.-R.; Boullerot, L.; Gamonet, C.; et al. Rapalogs Efficacy Relies on the Modulation of Antitumor T-cell Immunity. Cancer Res. 2016, 76, 4100–4112. [Google Scholar] [CrossRef] [Green Version]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Pedicord, V.A.; Cross, J.R.; Montalvo-Ortiz, W.; Miller, M.L.; Allison, J.P. Friends not foes: CTLA-4 blockade and mTOR inhibition cooperate during CD8+ T cell priming to promote memory formation and metabolic readiness. J. Immunol. 2015, 194, 2089–2098. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Phase 1b Neoadjuvant Trial of Sirolimus Followed by Durvalumab (MEDI4736) in Resectable Non-small Cell Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04348292 (accessed on 28 September 2021).
- Chouaib, S.; Umansky, V.; Kieda, C. The role of hypoxia in shaping the recruitment of proangiogenic and immunosuppressive cells in the tumor microenvironment. Wspolczesna Onkol. 2018, 22, 7–13. [Google Scholar] [CrossRef]
- Serra, H.; Chivite, I.; Angulo-Urarte, A.; Soler, A.; Sutherland, J.D.; Arruabarrena-Aristorena, A.; Ragab, A.; Lim, R.; Malumbres, M.; Fruttiger, M.; et al. PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat. Commun. 2015, 6, 7935. [Google Scholar] [CrossRef] [Green Version]
- Sopo, M.; Anttila, M.; Hämäläinen, K.; Kivelä, A.; Ylä-Herttuala, S.; Kosma, V.-M.; Keski-Nisula, L.; Sallinen, H. Expression profiles of VEGF-A, VEGF-D and VEGFR1 are higher in distant metastases than in matched primary high grade epithelial ovarian cancer. BMC Cancer 2019, 19, 584. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, P.E.; Mak, J.; Hernandez, G.; Jesudason, R.; Herault, A.; Javinal, V.; Borneo, J.; Kim, J.M.; Walsh, K.B. Anti-VEGF Treatment Enhances CD8+ T-cell Antitumor Activity by Amplifying Hypoxia. Cancer Immunol. Res. 2020, 8, 806–818. [Google Scholar] [CrossRef] [Green Version]
- ClinicaTrials.gov. Halting Early Advancement of Residual Disease by Treatment with Bevacizumab and Atezolizumab in Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04510584 (accessed on 20 October 2021).
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS ONE 2014, 9, e89350, Erratum in 2017, 12, e0186965. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. NSGO-OV-UMB1; ENGOT-OV30/NSGO: A Phase II Umbrella Trial in Patients with Relapsed Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03267589 (accessed on 28 September 2021).
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, Q.; Lian, X.; Jiang, P.; Cui, J. Hypoxia-Inducible Factor-1α (HIF-1α) Promotes Hypoxia-Induced Invasion and Metastasis in Ovarian Cancer by Targeting Matrix Metallopeptidase 13 (MMP13). Med. Sci. Monit. 2019, 25, 7202–7208. [Google Scholar] [CrossRef]
- Han, S.; Huang, T.; Hou, F.; Yao, L.; Wang, X.; Wu, X. The prognostic value of hypoxia-inducible factor-1α in advanced cancer survivors: A meta-analysis with trial sequential analysis. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875851. [Google Scholar] [CrossRef]
- Su, S.; Dou, H.; Wang, Z.; Zhang, Q. Bufalin inhibits ovarian carcinoma via targeting mTOR/HIF-α pathway. Basic Clin. Pharmacol. Toxicol. 2021, 128, 224–233. [Google Scholar] [CrossRef]
- Ai, Z.; Lu, Y.; Qiu, S.; Fan, Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016, 373, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Li, Y.; Lu, Y.; Wang, M.; Li, Y.; Wang, C.; Li, Q.; Zhao, H. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ 2021, 9, e11306. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e5. [Google Scholar] [CrossRef] [Green Version]
- Selnø, A.T.H.; Schlichtner, S.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Klenova, E.; Pavlova, L.; Gibbs, B.F.; Degen, M.; et al. Transforming growth factor beta type 1 (TGF-β) and hypoxia-inducible factor 1 (HIF-1) transcription complex as master regulators of the immunosuppressive protein galectin-9 expression in human cancer and embryonic cells. Aging 2020, 12, 23478. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumour microenvironment in PD-L1/PD-1-mediated immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced: MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Krasner, C.N.; Birrer, M.J.; Berlin, S.T.; Horowitz, N.S.; Buss, M.K.; Eliasof, S.; Garmey, E.G.; Hennessy, M.G.; Konstantinopoulos, P.; Matulonis, U. Phase II clinical trial evaluating CRLX101 in recurrent ovarian, tubal, and peritoneal cancer. J. Clin. Oncol. 2014, 32, 5581. [Google Scholar] [CrossRef]
- Krasner, C.N.; Campos, S.M.; Young, C.L.; Chadda, K.R.; Lee, H.; Birrer, M.J.; Horowitz, N.S.; Konstantinopoulos, P.A.; D’Ascanio, A.M.; Matulonis, U.A.; et al. Sequential Phase II clinical trials evaluating CRLX101 as monotherapy and in combination with bevacizumab in recurrent ovarian cancer. Gynecol. Oncol. 2021, 162, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Yoon, S.H.; Jang, H.; Jeong, J.H.; Lee, Y.M. Decursin promotes HIF-1α proteasomal degradation and immune responses in hypoxic tumour microenvironment. Phytomedicine 2020, 78, 153318. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. An Open-label, Randomized Phase 3 Study to Evaluate Efficacy and Safety of Pembrolizumab (MK-3475) in Combination with Belzutifan (MK-6482) and Lenvatinib (MK-7902), or MK-1308A in Combination with Lenvatinib, Versus Pembrolizumab and Lenvatinib, as First-Line Treatment in Participants with Advanced Clear Cell Renal Cell Carcinoma (ccRCC). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04736706 (accessed on 28 September 2021).
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.-M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 2016, 7, 41637–41649. [Google Scholar] [CrossRef] [Green Version]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [Green Version]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Haikala, H.M.; Anttila, J.; Klefström, J. MYC and AMPK-Save Energy or Die! Front. Cell Dev. Biol. 2017, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Wu, S.; Fukumoto, T.; Lin, J.; Nacarelli, T.; Wang, Y.; Ong, D.; Liu, H.; Fatkhutdinov, N.; Zundell, J.A.; Karakashev, S.; et al. Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat. Cancer 2021, 2, 189–200. [Google Scholar] [CrossRef]
- Macrophage Arginase Promotes Tumor Cell Growth and Suppresses Nitric Oxide-mediated Tumor Cytotoxicity|Cancer Research. Available online: https://cancerres.aacrjournals.org/content/61/3/1100.long (accessed on 23 October 2021).
- Miguel, R.D.V.; Cherpes, T.L.; Watson, L.J.; McKenna, K.C. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J. Immunol. 2010, 185, 6706–6718. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Safety, Pharmacokinetics, and Pharmacodynamics of Escalating Oral Doses of the Arginase Inhibitor INCB001158 (Formerly Known as CB1158) as a Single Agent and in Combination with Immune Checkpoint Therapy in Patients with Advanced/Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02903914 (accessed on 28 September 2021).
- Naing, A.; Bauer, T.; Papadopoulos, K.P.; Rahma, O.; Tsai, F.; Garralda, E.; Naidoo, J.; Pai, S.; Gibson, M.K.; Rybkin, I.; et al. Phase I study of the arginase inhibitor INCB001158 (1158) alone and in combination with pembrolizumab (PEM) in patients (Pts) with advanced/metastatic (adv/met) solid tumours. Ann. Oncol. 2019, 30, v160. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Lin, H.; Yuan, L.; Li, B. Combination therapy with L-arginine and α-PD-L1 antibody boosts immune response against osteosarcoma in immunocompetent mice. Cancer Biol. Ther. 2017, 18, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Nasreddine, G.; El-Sibai, M.; Abi-Habib, R.J. Cytotoxicity of [HuArgI (co)-PEG5000]-induced arginine deprivation to ovarian Cancer cells is autophagy dependent. Investig. New Drugs 2020, 38, 10–19. [Google Scholar] [CrossRef]
- Werner, A.; Koschke, M.; Leuchtner, N.; Luckner-Minden, C.; Habermeier, A.; Rupp, J.; Heinrich, C.; Conradi, R.; Closs, E.I.; Munder, M. Reconstitution of T Cell Proliferation under Arginine Limitation: Activated Human T Cells Take Up Citrulline via L-Type Amino Acid Transporter 1 and Use It to Regenerate Arginine after Induction of Argininosuccinate Synthase Expression. Front. Immunol. 2017, 8, 864. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Carvajal, R.D.; Pandit-Taskar, N.; Jungbluth, A.A.; Hoffman, E.W.; Wu, B.W.; Bomalaski, J.S.; Venhaus, R.; Pan, L.; Old, L.J.; et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Investig. New Drugs 2013, 31, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-Y.; Chiang, N.-J.; Wu, S.-Y.; Yen, C.-J.; Chen, S.-H.; Yeh, Y.-M.; Li, C.-F.; Feng, X.; Wu, K.; Johnston, A.; et al. Phase 1b study of pegylated arginine deiminase (ADI-PEG 20) plus Pembrolizumab in advanced solid cancers. Oncoimmunology 2021, 10, 1943253. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.X.; Cochrane, D.R.; Tessier-Cloutier, B.; Chen, S.Y.; Ho, G.; Pathak, K.V.; Alcazar, I.N.; Farnell, D.; Leung, S.; Cheng, A.; et al. Arginine Depletion Therapy with ADI-PEG20 Limits Tumor Growth in Argininosuccinate Synthase-Deficient Ovarian Cancer, Including Small-Cell Carcinoma of the Ovary, Hypercalcemic Type. Clin. Cancer Res. 2020, 26, 4402–4413. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Ricca, J.; Van Oudenhove, E.; Olvera, N.; Merghoub, T.; Levine, D.A.; Zamarin, D. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. J. Natl. Cancer Inst. 2018, 110, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. Tryptophan catabolism and T-cell tolerance: Immunosuppression by starvation? Immunol. Today 1999, 20, 469–473. [Google Scholar] [CrossRef]
- Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.S.; Firek, A.F.; Langridge, W.H.R. The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines 2015, 3, 703–729. [Google Scholar] [CrossRef] [Green Version]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [Green Version]
- Tanizaki, Y.; Kobayashi, A.; Toujima, S.; Shiro, M.; Mizoguchi, M.; Mabuchi, Y.; Yagi, S.; Minami, S.; Takikawa, O.; Ino, K. Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 2014, 105, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Saga, Y.; Mizukami, H.; Sato, N.; Nonaka, H.; Fujiwara, H.; Takei, Y.; Machida, S.; Takikawa, O.; Ozawa, K.; et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int. J. Oncol. 2012, 40, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Friberg, M.; Jennings, R.; Alsarraj, M.; Dessureault, S.; Cantor, A.; Extermann, M.; Mellor, A.L.; Munn, D.H.; Antonia, S.J. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101, 151–155. [Google Scholar] [CrossRef]
- Huang, T.T.; Yen, M.C.; Lin, C.C.; Weng, T.Y.; Chen, Y.L.; Lin, C.M.; Lai, M.D. Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci. 2011, 102, 2214–2220. [Google Scholar] [CrossRef]
- Yen, M.C.; Lin, C.C.; Chen, Y.L.; Huang, S.S.; Yang, H.J.; Chang, C.P.; Lei, H.Y.; Lai, M.D. A novel cancer therapy by skin delivery of indoleamine 2,3-dioxygenase siRNA. Clin. Cancer Res. 2009, 15, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Cushman, T.R.; Caetano, M.S.; Wang, X.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Li, G.; et al. Indoleamine 2,3-dioxygenase 1 inhibition targets anti-PD1-resistant lung tumors by blocking myeloid-derived suppressor cells. Cancer Lett. 2018, 431, 54–63. [Google Scholar] [CrossRef]
- Komiya, T.; Huang, C.H. Updates in the clinical development of Epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Zheng, X.; Koropatnick, J.; Chen, D.; Velenosi, T.; Ling, H.; Zhang, X.; Jiang, N.; Navarro, B.; Ichim, T.E.; Urquhart, B.; et al. Silencing IDO in dendritic cells: A novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int. J. Cancer 2012, 132, 967–977. [Google Scholar] [CrossRef]
- Deuster, E.; Mayr, D.; Hester, A.; Kolben, T.; Zeder-Göß, C.; Burges, A.; Mahner, S.; Jeschke, U.; Trillsch, F.; Czogalla, B. Correlation of the Aryl Hydrocarbon Receptor with FSHR in Ovarian Cancer Patients. Int. J. Mol. Sci. 2019, 20, 2862. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase–Mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758. [Google Scholar] [CrossRef]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. An Open-Label, Phase 1, First-in-Human, Dose Escalation and Expansion Study to Evaluate the Safety, Tolerability, Maximum Tolerated or Administered Dose, Pharmacokinetics, Pharmacodynamics and Tumor Response Profile of the Aryl Hydrocarbon Receptor Inhibitor (AhRi) BAY 2416964 in Participants with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04069026?term=BAY+2416964&cond=cancer&draw=2&rank=2 (accessed on 28 September 2021).
- ClinicalTrials.gov. An Open-Label, Phase 1b, Dose Escalation and Expansion Study to Evaluate the Safety, Tolerability, Maximum Tolerated or Administered Dose, Pharmacokinetics, Pharmacodynamics and Efficacy of the Aryl Hydrocarbon Receptor Inhibitor (AhRi) BAY 2416964 in Combination with Pembrolizumab in Participants with Advanced Solid Tumors. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04999202 (accessed on 28 September 2021).
- ClinicalTrials.gov. A Phase 1a/b, Open-Label, Dose-Escalation and Expansion Study of IK-175, an Oral Aryl Hydrocarbon Receptor (AHR) Inhibitor, as a Single Agent and in Combination With Nivolumab, a PD-1 Checkpoint Inhibitor in Patients with Locally Advanced or Metastatic Solid Tumors and Urothelial Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04200963 (accessed on 28 September 2021).
- Sadik, A.; Somarribas Patterson, L.F.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Boulland, M.-L.; Marquet, J.; Molinier-Frenkel, V.; Möller, P.; Guiter, C.; Lasoudris, F.; Copie-Bergman, C.; Baia, M.; Gaulard, P.; Leroy, K.; et al. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood 2007, 110, 220–227. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, W.; Liang, J.; Guo, J.; Ji, J.; Yao, Y.; Zheng, M.; Cai, Z.; Lu, L.; Wang, J. IL4I1 Is a Novel Regulator of M2 Macrophage Polarization That Can Inhibit T Cell Activation via L-Tryptophan and Arginine Depletion and IL-10 Production. PLoS ONE 2015, 10, e0142979. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Impact of the IL4I1 Enzyme Expression in Patients with Cutaneous Melanoma: Prognostic Value and/or Role in Resistance to Current Immunotherapy and Targeted Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT04253080 (accessed on 28 September 2021).
- Gonzalez Martin, A.; Sanchez Lorenzo, L.; Colombo, N.; Depont Christensen, R.; Heitz, F.; Meirovitz, M.; Selle, F.; Van Gorp, T.; Alvarez, N.; Sanchez, J.; et al. A phase III, randomized, double blinded trial of platinum based chemotherapy with or without atezolizumab followed by niraparib maintenance with or without atezolizumab in patients with recurrent ovarian, tubal, or peritoneal cancer and platinum treatment. Int. J. Gynecol. Cancer 2020, 31, 617–622. [Google Scholar] [CrossRef]
- Harter, P.; Pautier, P.; Van Nieuwenhuysen, E.; Reuss, A.; Redondo, A.; Lindemann, K.; Kurzeder, C.; Petru, E.; Heitz, F.; Sehouli, J.; et al. Atezolizumab in combination with bevacizumab and chemotherapy versus bevacizumab and chemotherapy in recurrent ovarian cancer—A randomized phase III trial (AGO-OVAR 2.29/ENGOT-ov34). Int. J. Gynecol. Cancer 2020, 30, 1997–2001. [Google Scholar] [CrossRef] [PubMed]
- Broekman, K.E.; Hof, M.A.J.; Touw, D.J.; Gietema, J.A.; Nijman, H.W.; Lefrandt, J.D.; Reyners, A.K.L.; Jalving, M. Phase I study of metformin in combination with carboplatin/paclitaxel chemotherapy in patients with advanced epithelial ovarian cancer. Investig. New Drugs 2020, 38, 1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Falcon, D.; Gregory-Goff, C.; Kinjyo, I.; Adams, S. Oophorectomy significantly impacts response to immune therapy regimens in preclinical models of ovarian cancer. Gynecol. Oncol. 2021, 162, S220. [Google Scholar] [CrossRef]
- Beura, L.K.; Hamilton, S.E.; Bi, K.; Schenkel, J.; Odumade, O.A.; Casey, K.A.; Thompson, E.A.; Fraser, K.A.; Rosato, P.C.; Filali-Mouhim, A.; et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016, 532, 512–516. [Google Scholar] [CrossRef]
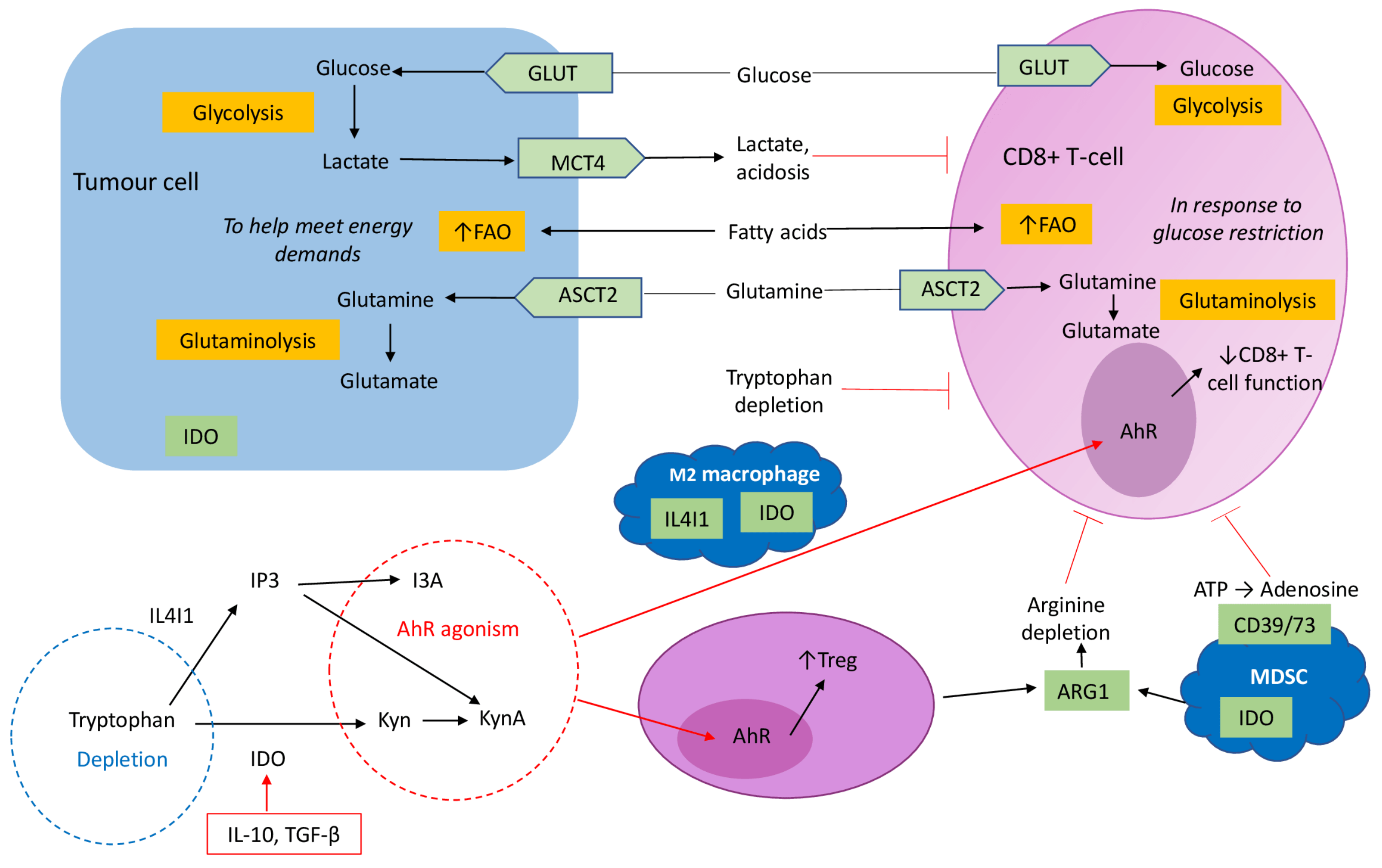


{kind=link}
{kind=link}
{kind=link}
| Study Phase and Design | Inclusion Criteria | No. of Patients | Treatment | NCT (ClinicalTrials.gov) |
|---|---|---|---|---|
| Phase I/II, open-label, sequential assignment Recruiting | AR-PR OC, triple negative breast, lung, prostate or colorectal carcinoma | 384 | Durvalumab + olaparib +/− cediranib (anti-VEGF) | NCT02484404 |
| Phase I/II, open-label, single group Active, not recruiting | RR OC/PPC/FTC with BRCA 1/2 mutation | 40 | Olaparib + tremelimumab + durvalumab | NCT02953457 |
| Phase II, triple masked, randomised Active, not recruiting | Recurrent PR OC/PPC/FTC | 122 | Atezolizumab + bevacizumab +/− placebo or acetylsalicylic acid | NCT02659384 |
| Phase II, open-label, single group assignment Recruiting | Recurrent PR HGSOC | 29 | Atezolizumab + bevacizumab + cobimetinib (mitogen-activated protein kinase inhibitor) | NCT03363867 |
| Phase II, open-label, single assignment Not yet recruiting | Recurrent PR OC/PPC/FTC or endometrial cancer | 47 | Lenvatinib + pembrolizumab + paclitaxel | NCT04781088 |
| Phase II, open-labelled, randomised Not yet recruiting | Recurrent OC/PPC/FTC with BRCA wild-type | 184 | Maintainence post platinum chemotherapy of olaparib +/− durvalumab +/− UV1 vaccine (hTERT) | NCT04742075 |
| Phase III, randomised, masked, parallel assignment Recruiting | Advanced epithelial OC with BRCA mutation | 1284 | First line treatment of carboplatin/paclitaxel + pembrolizumab or placebo Followed by maintenance of olaparib or placebo | NCT03740165 |
| Phase III, randomized, double blinded, placebo controlled Active, not recruiting | Stage III/IV, high grade non-mucinous epithelial OC/PPC/FTC | 1405 | Carboplatin/paclitaxel + bevacizumab; + placebo Or + dostarlimab (anti-PD-1) Or + niraparib | NCT03602859 |
| Phase III, randomized, double blinded, placebo controlled Active, not yet recruiting | Stage III/IV EOC/PPC/FTC who have completed cytoreductive surgery | 1000 | Maintenance post primary platinum-based chemotherapy; + nivolumab and rucaparib Or + nivolumab or placebo Or + rucaparib or placebo | NCT03522246 |
| Phase III, randomised, double-blinded Recruiting | Recurrent high-grade serous or endometroid OC/PPC/FTC | 414 | Carboplatin/paclitaxel + atezolizumab or placebo Followed by niraparib maintenance + atezolizumab or placebo | NCT03598270 |
| Phase III, randomised, parallel assignment Recruiting | Recurrent OC/PPC/FTC | 664 | Chemotherapy + bevacizumab + atezolizumab or placebo | NCT03353831 |
| Phase III, randomised, parallel assignment Recruiting | Recurrent, high-grade, PR OC | 444 | Doxorubicin +/− atezolizumab +/− bevacizumab | NCT02839707 |
| Phase III, randomised, double-blinded, placebo Recruiting | Advanced (III/IV) high-grade epithelial OC/PPC/FTC | 1374 | Platinum-based chemotherapy after primary/interval cytoreductive surgery and bevacizumab, followed by maintenance bevacizumab +/− durvalumab or placebo +/− olaparib or placebo | NCT03737643 |
| Study Phase and Design | CAR Target | Eligible | No. of Patients | Treatment | NCT |
|---|---|---|---|---|---|
| Interventional open-label single group Recruiting | Anti-mesothelin | RR OC with mesothelin positive tumour | 10 20 | Cyclophosphamide + Fludarabine With CAR T-cells | NCT03814447 |
| Interventional open-label single group Recruiting | Anti-mesothelin | RR OC with mesothelin positive tumour | 20 | CAR T-cells | NCT03916679 |
| Phase 1 open-label single group Recruiting | Anti-mesothelin | RR OC with mesothelin positive tumour | 34 | Cyclophosphamide + Fludarabine With CAR T-cells | NCT04562298 |
| Phase 1 open-label single group Recruiting | Anti-B7-H3 antigen | RR OC | 21 | Cyclophosphamide+ Fludarabine With CAR T-cells | NCT04670068 |
| Phase 1 open-label single group Recruiting | Anti-MUC16 (gene encoding ca 125) | RR OC/PPC/FTC | 71 | Biological: PRGN-3005 UltraCAR T-cells | NCT03907527 |
| Phase 1 open-label single group Not yet recruiting | Anti-ALPP | Metastatic ALPP positive OC and EC | 20 | CAR T-cells | NCT04627740 |
| Phase 1 open-label single group Recruiting | Anti-α-FR | RR HGSOC/PPC/FTC with α-FR positive tumour | 18 | CAR T-cells with or without Cyclophosphamide + Fludarabine | NCT03585764 |
| Exploratory open-label single group Recruiting | Anti-mesothelin T cells secreting PD-1 nanobodies | Mesothelin positive advanced solid tumours | 10 | CAR T-cells | NCT04503980 |
| Interventional open-label single group Recruiting | Autogolous Immunogene-modified T-Cells (IgT) | Stage III/IV OC in complete remission post primary treatment | 100 | CAR T-cells | NCT03184753 |
| Phase I open-label Recruiting | Anti-MUC1 | Advanced MUC1+ solid tumours (refractory OC) | 112 | Cyclophosphamide + Fludarabine With CAR T-cells | NCT04025216 |
| Study Phase and Design | Eligible | No. of Patients | Controls | NCT |
|---|---|---|---|---|
| Phase I open-label single-armActive, not yet recruiting | IIIc/IV OC no residual disease post primary treatment | 19 | Folate receptor alpha loaded DCV only | NCT02111941 |
| Phase I open-label single-armActive, not yet recruiting | HGSOC (=/>IIIb) post primary cytoreductive surgery + chemotherapy | 17 | DCV only | NCT04739527 |
| Phase I/IIa open-label single-arm Active, not yet recruiting | Stage II-IV OC no residual disease post primary treatment | 18 | Alpha-type-1 polarised DCV and intra-peritoneal infusion of CTL | NCT03735589 |
| Phase II Open-label single-arm Active, not yet recruiting | First recurrence of platinum-sensitive OC | 33 | Autologous maintenance DCV after standard chemotherapy | NCT03657966 |
| Phase II open-label single-arm Recruiting | AR OC | 36 | Autologous DCV only loaded with tumour lysate or for patients who are HLA-A2 with peptides of MUC1 and WT1 therapy | NCT00703105 |
| Phase II Multicentre, randomised, double-blind, placebo-controlled Recruiting | Stage III/IV OC/PPC no residual disease post primary treatment | 99 | Autologous DCV only loaded with tumour antigen versus loaded with peripheral blood mononuclear Cells | NCT02033616 |
| Phase II open-label randomised Active, not yet recruiting | AR OC | 23 | Autologous DCV plus GM-CSF | NCT00799110 |
| Phase III Multicentre, randomised, double-blind, placebo-controlled Active, not yet recruiting | AR platinum-sensitive OC | 678 | Induction: DCV verus placebo with carboplatin + gemcitabine/paclitaxel/doxorubicin +/− bevacizumab Maintenance: DCV versus placebo + bevacizumab +/− PARPi | NCT03905902 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, R.L.; Cummings, M.; Thangavelu, A.; Theophilou, G.; de Jong, D.; Orsi, N.M. Barriers to Immunotherapy in Ovarian Cancer: Metabolic, Genomic, and Immune Perturbations in the Tumour Microenvironment. Cancers 2021, 13, 6231. https://doi.org/10.3390/cancers13246231
Johnson RL, Cummings M, Thangavelu A, Theophilou G, de Jong D, Orsi NM. Barriers to Immunotherapy in Ovarian Cancer: Metabolic, Genomic, and Immune Perturbations in the Tumour Microenvironment. Cancers. 2021; 13(24):6231. https://doi.org/10.3390/cancers13246231
Chicago/Turabian StyleJohnson, Racheal Louise, Michele Cummings, Amudha Thangavelu, Georgios Theophilou, Diederick de Jong, and Nicolas Michel Orsi. 2021. "Barriers to Immunotherapy in Ovarian Cancer: Metabolic, Genomic, and Immune Perturbations in the Tumour Microenvironment" Cancers 13, no. 24: 6231. https://doi.org/10.3390/cancers13246231
APA StyleJohnson, R. L., Cummings, M., Thangavelu, A., Theophilou, G., de Jong, D., & Orsi, N. M. (2021). Barriers to Immunotherapy in Ovarian Cancer: Metabolic, Genomic, and Immune Perturbations in the Tumour Microenvironment. Cancers, 13(24), 6231. https://doi.org/10.3390/cancers13246231