
Drug Repositioning and Subgroup Discovery for Precision Medicine Implementation in Triple Negative Breast Cancer

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
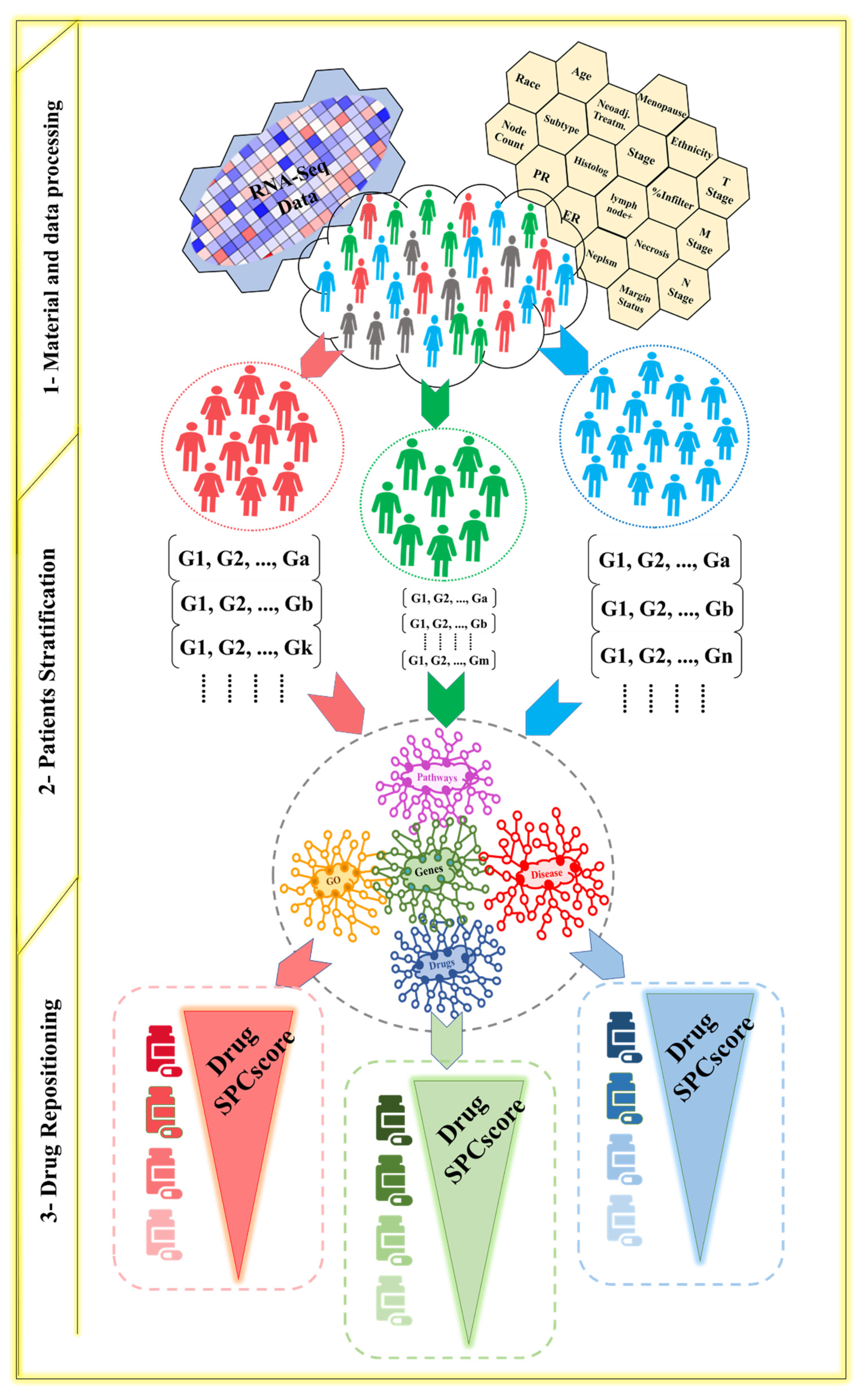
2. Materials and Methods
2.1. Patient Stratification
2.2. Drug Repositioning
3. Results
3.1. Subgroups and Drugs Analysis
- Subgroup1 is for patients who are Black or African-American with no history of a neoadjuvant treatment. This subgroup has 30 patients, and it has 286 unique DEGs to this group (Tables S3 and S4). The drugs that can induce ferroptosis that were suggested for this subgroup are Afatinib, Gefitinib, Bosutinib, Lapatinib, and Fulvestrant.
- Afatinib was found to be a ferroptosis inducer in TNBC by targeting EGFR [59]. Afatinib’s mechanism of action includes binding to wild-type epidermal growth factor receptors (EGFR) and irreversibly inhibiting the kinase activity of all ErbB family members, which are well-recognized as an oncogenic driver in epithelial cancers. Thus, Afatinib can inhibit the proliferation of cancer cell lines [60]. Analyzing the patient profiles in this subgroup showed that EGFR was abnormally expressed in this subgroup. ErbB4 had an abnormal expression as well.
- Gefitinib is an epidermal growth factor receptor (EGFR) and a tyrosine kinase inhibitor. It has antiproliferative and anti-tumoral activity in BC [61,62]. Gefitinib targets EGFR and is used to induce ferroptosis [59]. The proliferation of TNBC cells can be inhibited by targeting EGFR kinase activity using gefitinib [63]. Moreover, it can be used as a combined therapy with multiple EGFR-TKIs, such as lapatinib and erlotinib, to overcome the resistance to EGFR-targeted therapy in TNBC. The inhibition of EGFR, in combination with the dual inhibition of cdc7/CDK9, results in reduced cell proliferation, accompanied by the induction of apoptosis, the G2-M cell cycle arrest, the inhibition of DNA replication, and the abrogation of CDK9-mediated transcriptional elongation in TNBC cells [63]. Using gefitinib for this subgroup will inhibit EGFR and cdc7 that was upregulated in this subgroup. Inhibiting them is necessary to reduce cell proliferation. CDK1 and CDK5, which are cyclin-dependent kinases (CDKs), were found to be upregulated in this subgroup. These protein kinases control cancer progression and inhibiting them prevents cancer proliferation [64].
- Bosutinib promotes autophagy and research has shown that there is crosstalk between autophagy and ferroptosis [65]. BCR-Abl is the target for bosutinib, dasatinib, imatinib, nilotinib, ponatinib, and regorafenib. Bosutinib is a dual Src/Abl inhibitor [66]. In this subgroup, Src was upregulated; therefore, this drug will inhibit Src and reduce cell proliferation. Src belongs to a different set of mechanisms associated with cancer progression, like inducing a metastatic phenotype, enhancing tumor growth, and enhancing angiogenesis [67].
- Lapatinib is a tyrosine kinase inhibitor. This drug was used as a combined therapy with siramesine, which is a lysosome disrupting agent, to induce ferroptosis and reactive oxygen species (ROS) in TNBC [68]. Lapatinib is an inhibitor of ErbB1 and ErbB2 and induces ferroptosis in BC cell lines by altering iron regulation. It is used as a combined therapy to induce ferroptosis in TNBC by inhibiting the iron transport system, leading to an increase in ROS and cell death [69]. Ferroportin-1 (FPN) is an iron transport protein responsible for the removal of iron from cells. Its expression decreases after treatment with siramesine in combination with lapatinib. The overexpression of FPN decreases ROS and cell death, whereas the knockdown of FPN increases cell death after siramesine and lapatinib treatment. This indicates a novel induction of ferroptosis through altered iron regulation by treating breast cancer cells with a lysosome disruptor and a tyrosine kinase inhibitor [68]. Lapatinib is a tyrosine kinase inhibitor of the epidermal growth factor receptor (EGFR) and ErbB2 (HER2) tyrosine kinases. Studies in vitro showed that lapatinib inhibited the proliferation of ErbB2 and EGFR, which are overexpressed in cancer cells [70,71]. Siramesine and lapatinib gave the best combination index, and this combination induced ferroptosis through iron-mediated ROS and the downregulation of heme oxygenase 1 (HO-1) levels [72]. In TNBC, siramesine and lapatinib increases Transferrin Receptor (TFRC) expression and decreases ferroportin1 (FPN1) expression, thus elevating the level of intracellular iron [73].
- Subgroup2: The patients in this subgroup are not Hispanic or Latino and White; they have less than three positive lymph nodes, and their histological type is an intraductal carcinoma. This subgroup has 37 patients and 87 unique DEGs that are abnormal in this subgroup, but not in the other subgroups (Tables S3 and S4). The drugs that can induce ferroptosis that are suggested for this subgroup are fluvastatin, lovastatin, and gefitinib.
- Fluvastatin targets HMG-CoA and can induce ferroptosis by decreasing the expression of glutathione peroxidase 4 (GPX4). This effect is time- and concentration-dependent [74,75]. Treatment with this statin induces ferroptosis by inhibiting GPX4 and the key products in the mevalonate pathway, like 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, and the depletion of CoQ10, that reduces the levels of this key membrane antioxidant [74,76,77]. Fluvastatin can be used as a combined therapy with RSL3, which is a direct inhibitor of GPX4 [74].
- Gefitinib: As mentioned previously, it can be used to induce ferroptosis in TNBC [59].
- c.
- Subgroup3: The patients in this subgroup are not Hispanic or Latino. They have right-sided TNBC as their neoplasm subdivision, they have less than three lymph nodes that are positive, and their histological type is an intraductal carcinoma. This subgroup contains 30 patients and has 118 unique DEGs that are abnormal in this subgroup, but not in the other subgroups (Tables S3 and S4). The drugs that can induce ferroptosis that are suggested for this subgroup are sunitinib and pazopanib.
- Pazopanib: Pazopanib belongs to the same category as Sunitinib. Moreover, treating breast cancer cells with Pazopanib was found to induce autophagic cell death [84]. It is a necroptosis inhibitor [85]. The mTOR pathway is a regulator of iron metabolism. The VHL/HIF-α axis is the main regulator target of iron metabolism. HIF-3α was downregulated in this subgroup. Targeting the VHL gene pathway using drugs like sunitinib, sorafenib, pazopanib, and axitinib causes the VHL to be inactive. The inactivation of the VHL increases sensitivity to ferroptosis. IRP1 can bind to the iron reaction element of HIF-2α mRNA and inhibit its translation. Tempol, an IRP1-activated drug, inhibits HIF-2α and HIF-1α protein levels. PT2399 and PT2385 are inhibitors of HIF-2α [86]. Pazopanib is a tyrosine kinase inhibitor (TKI) that belongs to a class of drugs that targets PDGFR α/β and VEGFR activity [66]. PDGFR was found to be downregulated in this subgroup. After analyzing the genes in this subgroup, we found that sunitinib and pazopanib bind to FGFR2, which, in turn, interacts with HSPB1 and STAT3. FGFR2 was upregulated in this subgroup. Both of these genes play a role in ferroptosis induction, as explained previously. Moreover, sunitinib binds to PHKG2, which is important for the induction of ferroptosis [87,88,89].
- d.
- Subgroup4: The patients in this subgroup are not Hispanic or Latino; they have left side TNBC as their Neoplasm Subdivision. They have less than three lymph nodes that are positive, and their histological type is an intraductal carcinoma. This subgroup has 32 patients and has 103 unique DEGs that are abnormal in this subgroup, but not in the other subgroups (Tables S3 and S4). The drugs that have the potential to induce ferroptosis in this subgroup are dexamethasone and vorinostat.
- Dexamethasone: High-dose dexamethasone disrupts the metabolism of glutamate and cysteine, produces more ROS, and downregulates GPX4 and system XC−, which are two key mediators of ferroptosis [90]. In this subgroup, ROS1 was upregulated, GPX2–3 was downregulated, GPX5 was upregulated, and GPX4 was in the normal range.
- Vorinostat: Vorinostat is a histone deacetylase (HDAC) inhibitor. In colon cancer cells, vorinostat can significantly inhibit cell growth and can induce cell cycle arrest and apoptosis [91]. It may induce ferroptosis by glutamine deprivation, resulting in the accumulation of ROS [92]. In NSCLC, vorinostat, combined with erastin, can increase the lipid peroxide levels and can inhibit HDAC to induce ferroptosis [93]. Moreover, when vorinostat is used as a combined therapy with gefitinib or erlotinib, which are EGFR-TKIs, it can promote oxidative stress-dependent apoptosis by the suppression of the c-MYC-regulated NRF2 functions and increase the levels of KEAP1 in NSCLC cells [94].
- e.
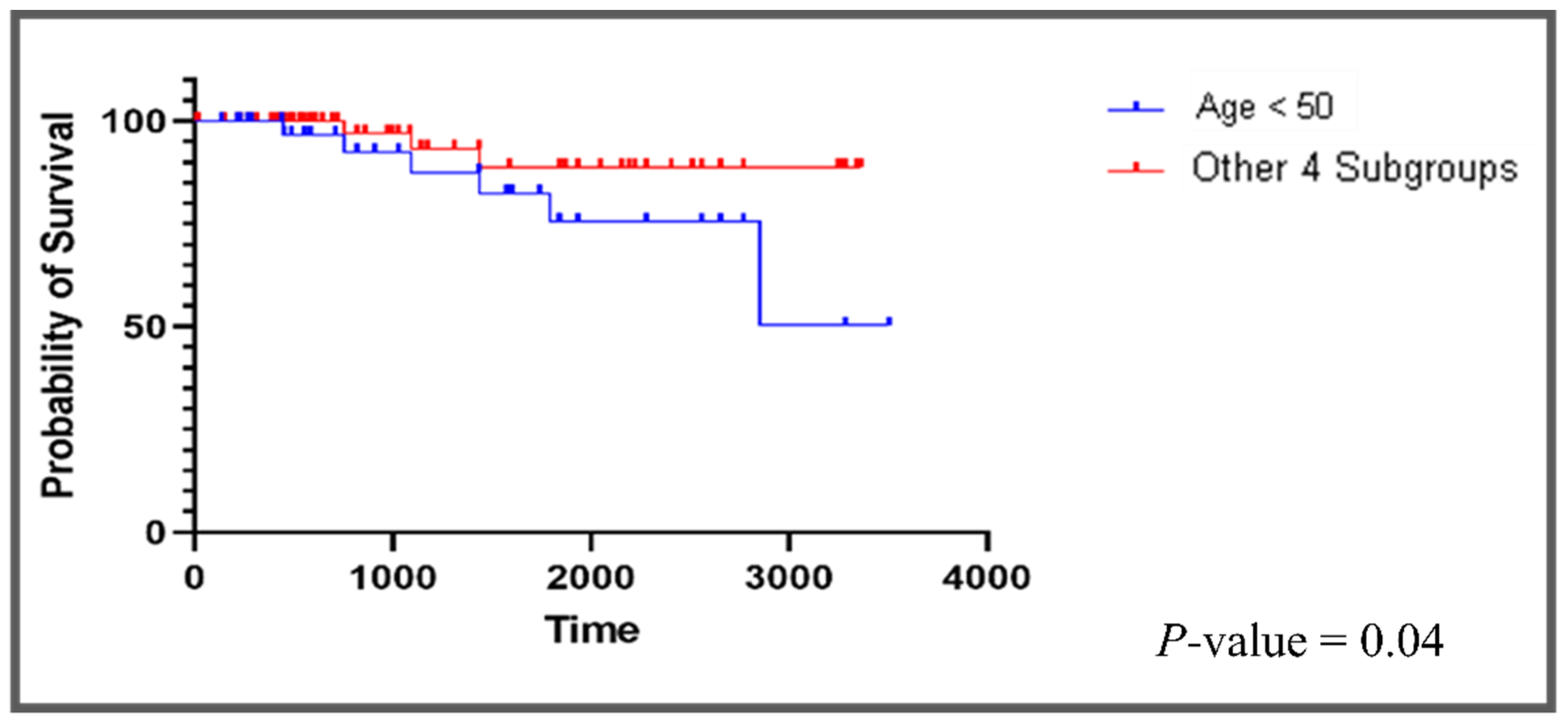
- Subgroup5: The patients in this subgroup are less than 50 years old; their pathological stage is m0 with no history of neoadjuvant treatment. This subgroup has 37 patients and has 216 unique DEGs that are abnormal in this subgroup, but not in the other subgroups (Tables S3 and S4). Opposite to what we observed in the other four subgroups, most of the top drugs that are suggested to this subgroup have an antioxidative effect on cancer cells. These drugs are rifampicin, galantamine, cerulenin, and lipoic acid.
- Galantamine: Galantamine has an antioxidant effect and acethylcholinesterase and γ-secretase inhibitory activity [97].
- Cerulenin: Cerulenin is an antifungal antibiotic that inhibits fatty acid and steroid biosynthesis. Fatty acid synthase (FAS) was observed to have a high expression in breast cancer cells in comparison with the normal cells, and there is increasing evidence that FAS plays a role in breast cancer development [98]. Moreover, fatty acid synthase (FAS) and ErbB2 have been shown to promote breast cancer cell migration [99]. The effect of cerulenin on breast cancer was tested in an in vivo analysis. Due to cerulenin’s ability to inhibit fatty acid synthase (FAS) that, in turn, decreases the expression of ErbB1, 2, and 4, it may initiate an epithelial-to-mesenchymal transition (EMT) as well as the migration and invasive ability of cancer cells [100].
- Lipoic acid (LA): Lipoic acid is an antioxidant. It has an anti-proliferative effect in breast cancer cells by reducing breast cancer cell viability, cell cycle progression, and the EMT. It downregulates furin, which, in turn, inhibits the maturation of IGF-1R [101,102]. Combining lipoic acid with radiation therapy was found to overcome the resistance to radiation therapy and promote apoptosis [103].
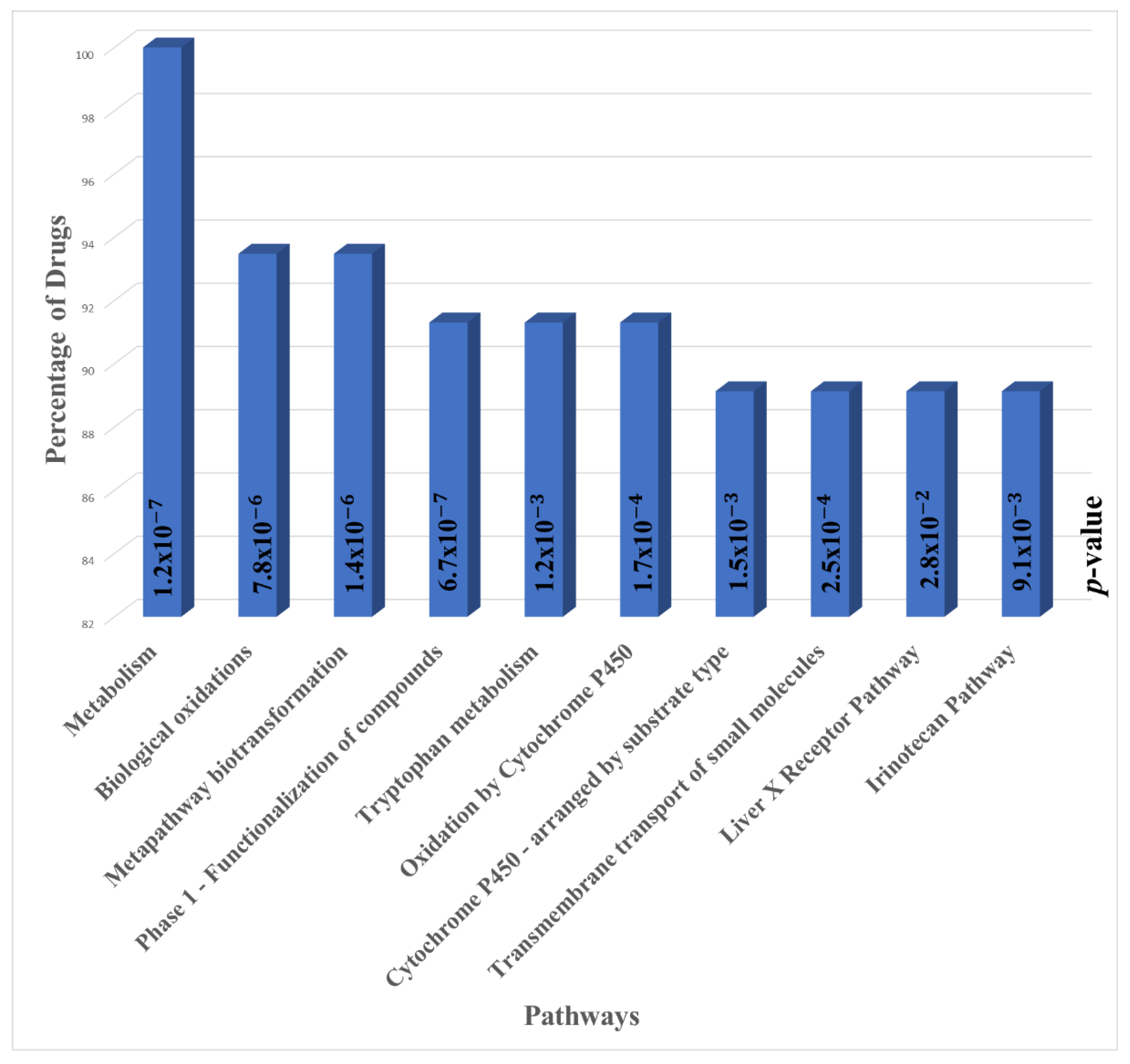
3.2. Pathway Enrichment Analysis
3.3. Pharmacological Classes Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kunnumakkara, A.B.; Bordoloi, D.; Sailo, B.L.; Roy, N.K.; Thakur, K.K.; Banik, K.; Shakibaei, M.; Gupta, S.C.; Aggarwal, B.B. Cancer drug development: The missing links. Exp. Biol. Med. 2019, 244, 663–689. [Google Scholar] [CrossRef]
- Hicks, S.C.; Okrah, K.; Paulson, J.N.; Quackenbush, J.; Irizarry, R.A.; Bravo, H.C. Smooth quantile normalization. Biostatistics 2018, 19, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor heterogeneity in breast cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Alaimo, S.; Pulvirenti, A. Network-based drug repositioning: Approaches, resources, and research directions. Methods Mol. Biol. 2019, 1903, 97–113. [Google Scholar] [CrossRef]
- Deotarse, P.; Jain, A.; Baile, M.; Kolhe, N.; Kulkarni, A. Drug repositioning: A review. Int. J. Pharma Res. Rev. 2015, 4, 51–58. [Google Scholar]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232. [Google Scholar] [CrossRef] [Green Version]
- Tian, K.; Bakker, E.; Hussain, M.; Guazzelli, A.; Alhebshi, H.; Meysami, P.; Demonacos, C.; Schwartz, J.-M.; Mutti, L.; Krstic-Demonacos, M. p53 modeling as a route to mesothelioma patients stratification and novel therapeutic identification. J. Transl. Med. 2018, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masi, M.; Garattini, E.; Bolis, M.; Di Marino, D.; Maraccani, L.; Morelli, E.; Grolla, A.A.; Fagiani, F.; Corsini, E.; Travelli, C. OXER1 and RACK1-associated pathway: A promising drug target for breast cancer progression. Oncogenesis 2020, 9, 1–15. [Google Scholar] [CrossRef]
- Jin, G.; Fu, C.; Zhao, H.; Cui, K.; Chang, J.; Wong, S.T. A novel method of transcriptional response analysis to facilitate drug repositioning for cancer therapy. Cancer Res. 2012, 72, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Louhimo, R.; Laakso, M.; Belitskin, D.; Klefstrom, J.; Lehtonen, R.; Hautaniemi, S. Data integration to prioritize drugs using genomics and curated data. BioData Min. 2016, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cava, C.; Bertoli, G.; Castiglioni, I. In silico identification of drug target pathways in breast cancer subtypes using pathway cross-talk inhibition. J. Transl. Med. 2018, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Carrella, D.; Manni, I.; Tumaini, B.; Dattilo, R.; Papaccio, F.; Mutarelli, M.; Sirci, F.; Amoreo, C.A.; Mottolese, M.; Iezzi, M. Computational drugs repositioning identifies inhibitors of oncogenic PI3K/AKT/P70S6K-dependent pathways among FDA-approved compounds. Oncotarget 2016, 7, 58743. [Google Scholar] [CrossRef] [Green Version]
- Turanli, B.; Karagoz, K.; Bidkhori, G.; Sinha, R.; Gatza, M.L.; Uhlen, M.; Mardinoglu, A.; Arga, K.Y. Multi-omic data interpretation to repurpose subtype specific drug candidates for breast cancer. Front. Genet. 2019, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Juan, Y.S.; Wu, M.S.; Zhang, Y.F.; Hsu, Y.W.; Chen, H.H.; Liu, W.M.; Chang, W.C. Integrative bioinformatic analyses of an oncogenomic profile reveal the biology of endometrial cancer and guide drug discovery. Oncotarget 2016, 7, 5909–5923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyvandipour, A.; Saberian, N.; Shafi, A.; Donato, M.; Draghici, S. A novel computational approach for drug repurposing using systems biology. Bioinformatics 2018, 34, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Meng, F.; Dai, E.; Yang, F.; Wang, S.; Chen, X.; Yang, L.; Wang, Y.; Jiang, W. Identification of associations between small molecule drugs and miRNAs based on functional similarity. Oncotarget 2016, 7, 38658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Li, Z.; Fan, X.; Cheng, Y. Drug–disease association and drug-repositioning predictions in complex diseases using causal inference–probabilistic matrix factorization. J. Chem. Inf. Modeling 2014, 54, 2562–2569. [Google Scholar] [CrossRef]
- Fahimian, G.; Zahiri, J.; Arab, S.S.; Sajedi, R.H. RepCOOL: Computational drug repositioning via integrating heterogeneous biological networks. J. Transl. Med. 2020, 18, 1–10. [Google Scholar] [CrossRef]
- Klahan, S.; Wong, H.S.; Tu, S.H.; Chou, W.H.; Zhang, Y.F.; Ho, T.F.; Liu, C.Y.; Yih, S.Y.; Lu, H.F.; Chen, S.C.; et al. Identification of genes and pathways related to lymphovascular invasion in breast cancer patients: A bioinformatics analysis of gene expression profiles. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Karuppasamy, R.; Verma, K.; Sequeira, V.M.; Basavanna, L.N.; Veerappapillai, S. An Integrative drug repurposing pipeline: Switching viral drugs to breast cancer. J. Cell. Biochem. 2017, 118, 1412–1422. [Google Scholar] [CrossRef]
- Rymbai, E.; Sugumar, D.; Saravanan, J.; Divakar, S. Ropinirole, a potential drug for systematic repositioning based on side effect profile for management and treatment of Breast Cancer. Med. Hypotheses 2020, 144, 110156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Jin, G.; Cui, K.; Ren, D.; Liu, T.; Chen, P.; Wong, S.; Li, F.; Fan, Y.; Rodriguez, A. Novel modeling of cancer cell signaling pathways enables systematic drug repositioning for distinct breast cancer metastases. Cancer Res. 2013, 73, 6149–6163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Zhao, J.; Gao, L. Predicting potential drugs for breast cancer based on miRNA and tissue specificity. Int. J. Biol. Sci. 2018, 14, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Jadamba, E.; Shin, M. A systematic framework for drug repositioning from integrated omics and drug phenotype profiles using pathway-drug network. BioMed Res. Int. 2016, 2016, 7147039. [Google Scholar] [CrossRef]
- Cava, C.; Sabetian, S.; Castiglioni, I. Patient-specific network for personalized breast cancer therapy with multi-omics data. Entropy 2021, 23, 225. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, J. Integration of a prognostic gene module with a drug sensitivity module to identify drugs that could be repurposed for breast cancer therapy. Comput. Biol. Med. 2015, 61, 163–171. [Google Scholar] [CrossRef]
- Johnson, K.S.; Conant, E.F.; Soo, M.S. Molecular subtypes of breast cancer: A review for breast radiologists. J. Breast Imaging 2021, 3, 12–24. [Google Scholar] [CrossRef]
- Cava, C.; Pisati, M.; Frasca, M.; Castiglioni, I. Identification of breast cancer subtype-specific biomarkers by integrating copy number alterations and gene expression profiles. Medicina 2021, 57, 261. [Google Scholar] [CrossRef]
- Low, Y.S.; Daugherty, A.C.; Schroeder, E.A.; Chen, W.; Seto, T.; Weber, S.; Lim, M.; Hastie, T.; Mathur, M.; Desai, M. Synergistic drug combinations from electronic health records and gene expression. J. Am. Med. Inform. Assoc. 2017, 24, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Bourdakou, M.M.; Athanasiadis, E.I.; Spyrou, G.M. Discovering gene re-ranking efficiency and conserved gene-gene relationships derived from gene co-expression network analysis on breast cancer data. Sci. Rep. 2016, 6, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, G.; Cava, C.; Castiglioni, I. The potential of miRNAs for diagnosis, treatment and monitoring of breast cancer. Scand. J. Clin. Lab. Investig. 2016, 76, S34–S39. [Google Scholar] [CrossRef]
- Sarkar, J.P.; Saha, I.; Sarkar, A.; Maulik, U. Machine learning integrated ensemble of feature selection methods followed by survival analysis for predicting breast cancer subtype specific miRNA biomarkers. Comput. Biol. Med. 2021, 131, 104244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, L.; Yang, Q.; Liu, H.; Meng, Q.; Jiang, L.; Wang, S.; Jiang, W. Systematical analysis of lncRNA–mRNA competing endogenous RNA network in breast cancer subtypes. Breast Cancer Res. Treat. 2018, 169, 267–275. [Google Scholar] [CrossRef]
- Mejía-Pedroza, R.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway-based drug repositioning for breast cancer molecular subtypes. Front. Pharmacol. 2018, 9, 905. [Google Scholar] [CrossRef] [Green Version]
- Warchal, S.J.; Dawson, J.C.; Shepherd, E.; Munro, A.F.; Hughes, R.E.; Makda, A.; Carragher, N.O. High content phenotypic screening identifies serotonin receptor modulators with selective activity upon breast cancer cell cycle and cytokine signaling pathways. Bioorg. Med. Chem. 2020, 28, 115209. [Google Scholar] [CrossRef]
- Schneider, L.; Kehl, T.; Thedinga, K.; Grammes, N.L.; Backes, C.; Mohr, C.; Schubert, B.; Lenhof, K.; Gerstner, N.; Daniel Hartkopf, A.; et al. ClinOmicsTrailbc: A visual analytics tool for breast cancer treatment stratification. Bioinformatics 2019, 35, 5171–5181. [Google Scholar] [CrossRef] [Green Version]
- Al-Taie, Z.; Liu, D.; Mitchem, J.B.; Papageorgiou, C.; Kaifi, J.T.; Warren, W.C.; Shyu, C.-R. Explainable artificial intelligence in high-throughput drug repositioning for subgroup stratifications with interventionable potential. J. Biomed. Inform. 2021, 118, 103792. [Google Scholar] [CrossRef]
- Metzger-Filho, O.; Tutt, A.; de Azambuja, E.; Saini, K.S.; Viale, G.; Loi, S.; Bradbury, I.; Bliss, J.M.; Azim, H.A., Jr.; Ellis, P.; et al. Dissecting the heterogeneity of triple-negative breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1879–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Kassam, F.; Enright, K.; Dent, R.; Dranitsaris, G.; Myers, J.; Flynn, C.; Fralick, M.; Kumar, R.; Clemons, M. Survival outcomes for patients with metastatic triple-negative breast cancer: Implications for clinical practice and trial design. Clin. Breast Cancer 2009, 9, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Klahan, S.; Huang, W.C.; Chang, C.M.; Wong, H.S.; Huang, C.C.; Wu, M.S.; Lin, Y.C.; Lu, H.F.; Hou, M.F.; Chang, W.C. Gene expression profiling combined with functional analysis identify integrin beta1 (ITGB1) as a potential prognosis biomarker in triple negative breast cancer. Pharmacol. Res. 2016, 104, 31–37. [Google Scholar] [CrossRef]
- Chang, W.C.; Woon, P.Y.; Hsu, Y.W.; Yang, S.; Chiu, Y.C.; Hou, M.F. The association between single-nucleotide polymorphisms of ORAI1 gene and breast cancer in a Taiwanese population. Sci. World J. 2012, 2012, 916587. [Google Scholar] [CrossRef] [Green Version]
- Klahan, S.; Wu, M.S.; Hsi, E.; Huang, C.C.; Hou, M.F.; Chang, W.C. Computational analysis of mRNA expression profiles identifies the ITG family and PIK3R3 as crucial genes for regulating triple negative breast cancer cell migration. BioMed Res. Int. 2014, 2014, 536591. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Anderson, B.O.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; et al. Breast Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. JNCCN 2020, 18, 452–478. [Google Scholar] [CrossRef] [Green Version]
- Himmelstein, D.S.; Lizee, A.; Hessler, C.; Brueggeman, L.; Chen, S.L.; Hadley, D.; Green, A.; Khankhanian, P.; Baranzini, S.E. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. Elife 2017, 6, e26726. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Baskett, W.; Beversdorf, D.; Shyu, C.-R. Exploratory data mining for subgroup cohort discoveries and prioritization. IEEE J. Biomed. Health Inform. 2019, 24, 1456–1468. [Google Scholar] [CrossRef]
- Agrawal, R.; Srikant, R. Fast algorithms for mining association rules. In Proceedings of the 20th International Conference Very Large Data Bases, VLDB, Santiago de Chile, Chile, 12–15 September 1994; pp. 487–499. [Google Scholar]
- Dong, G.; Li, J. Efficient mining of emerging patterns: Discovering trends and differences. In Proceedings of the Fifth ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Diego, CA, USA, 15–18 August 1999; pp. 43–52. [Google Scholar]
- Hoeting, J.A.; Madigan, D.; Raftery, A.E.; Volinsky, C.T. Correction to: “Bayesian model averaging: A tutorial” [Statist. Sci. 14 (1999), no. 4, 382-417; MR 2001a: 62033]. Stat. Sci. 2000, 15, 193–195. [Google Scholar] [CrossRef]
- Hoeting, J.A. Methodology for Bayesian model averaging: An update. In Proceedings of the Proceedings-Manuscripts of Invited Paper Presentations, International Biometric Conference, Freiburg, Germany, 21–26 July 2002; pp. 231–240. [Google Scholar]
- Hoeting, J.A.; Madigan, D.; Raftery, A.E.; Volinsky, C.T. Bayesian model averaging: A tutorial (with comments by M. Clyde, David Draper and EI George, and a rejoinder by the authors. Stat. Sci. 1999, 14, 382–417. [Google Scholar] [CrossRef]
- Church, K.; Gale, W. Inverse document frequency (idf): A measure of deviations from poisson. In Natural Language Processing Using Very Large Corpora; Springer: Berlin/Heidelberg, Germany, 1999; pp. 283–295. [Google Scholar]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular classification and molecular forecasting of breast cancer: Ready for clinical application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Abramson, V.; Tan, T.; Dent, R. The evolution of triple-negative breast cancer: From biology to novel therapeutics. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, 34–42. [Google Scholar] [CrossRef]
- Jhan, J.-R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef] [Green Version]
- Vitali, F.; Cohen, L.D.; Demartini, A.; Amato, A.; Eterno, V.; Zambelli, A.; Bellazzi, R. A network-based data integration approach to support drug repurposing and multi-target therapies in triple negative breast cancer. PLoS ONE 2016, 11, e0162407. [Google Scholar]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Q.; Zuo, Z.X.; Yuan, S.Q.; Yu, K.; Zhang, Q.; Zhang, X.; Sheng, H.; Ju, H.Q.; Cheng, H.; et al. Systematic analysis of the aberrances and functional implications of ferroptosis in cancer. Iscience 2020, 23, 101302. [Google Scholar] [CrossRef] [PubMed]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Bernsdorf, M.; Ingvar, C.; Jörgensen, L.; Tuxen, M.K.; Jakobsen, E.H.; Saetersdal, A.; Kimper-Karl, M.L.; Kroman, N.; Balslev, E.; Ejlertsen, B. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res. Treat. 2011, 126, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Gründker, C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol. Rep. 2017, 37, 1212–1218. [Google Scholar] [CrossRef]
- McLaughlin, R.P.; He, J.; Van Der Noord, V.E.; Redel, J.; Foekens, J.A.; Martens, J.W.; Smid, M.; Zhang, Y.; Van de Water, B. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019, 21, 1–15. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclins and related kinases in cancer cells. J. BU ON. Off. J. Balk. Union Oncol. 2007, 12, S45–S52. [Google Scholar]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630. [Google Scholar]
- Smidova, V.; Michalek, P.; Goliasova, Z.; Eckschlager, T.; Hodek, P.; Adam, V.; Heger, Z. Nanomedicine of tyrosine kinase inhibitors. Theranostics 2021, 11, 1546. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Henson, E.; Chen, Y.; Gibson, S. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar] [PubMed]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [Green Version]
- Villalpando-Rodriguez, G.E.; Blankstein, A.R.; Konzelman, C.; Gibson, S.B. Lysosomal destabilizing drug siramesine and the dual tyrosine kinase inhibitor lapatinib induce a synergistic ferroptosis through reduced heme oxygenase-1 (HO-1) levels. Oxidative Med. Cell. Longev. 2019, 2019, 9561281. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, L.; Chen, C.; Zhou, Y.; Hu, D.; Yang, J.; Chen, Y.; Zhuo, W.; Mao, M.; Zhang, X. Targeting ferroptosis in breast cancer. Biomark. Res. 2020, 8, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, L.; Peng, X.; Fan, Q.; Wei, S.; Yang, S.; Li, X.; Jin, H.; Wu, B.; Huang, M. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomed. Pharmacother. 2020, 130, 110710. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-F.; Kim, D.-H.; Yoon, Y.-S.; Kim, S.-K.; Cai, D.-Q.; Teng, Y.-C.; Shim, K.-Y.; Lee, K.-J. Involvement of oxidative stress in simvastatin-induced apoptosis of murine CT26 colon carcinoma cells. Toxicol. Lett. 2010, 199, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The hallmarks of ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Wu, Y.; Yu, C.; Luo, M.; Cen, C.; Qiu, J.; Zhang, S.; Hu, K. Ferroptosis in cancer treatment: Another way to Rome. Front. Oncol. 2020, 10, 571127. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-dependent ferroptosis: Machinery and regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Chen, J.J.; Galluzzi, L. Fighting resilient cancers with iron. Trends Cell Biol. 2018, 28, 77–78. [Google Scholar] [CrossRef]
- Lin, X.; Ping, J.; Wen, Y.; Wu, Y. The mechanism of ferroptosis and applications in tumor treatment. Front. Pharmacol. 2020, 11, 1061. [Google Scholar] [CrossRef]
- Lai, Y.; Zeng, T.; Liang, X.; Wu, W.; Zhong, F.; Wu, W. Cell death-related molecules and biomarkers for renal cell carcinoma targeted therapy. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Farfariello, V.; Nabissi, M.; Liberati, S.; Bonfili, L.; Mozzicafreddo, M.; Eleuteri, A.M.; Burattini, L. Different Effects of Sunitinib, Sorafenib, and Pazopanib on Inducing Cancer Cell Death: The Role of Autophagy. In Proceedings of the Genitourinary Cancers Symposium, Orlando, FL, USA, 14–16 February 2013; American Society of Clinical Oncology: Alexandria, VA, USA, 2013; p. 270. [Google Scholar]
- Fulda, S. Repurposing anticancer drugs for targeting necroptosis. Cell Cycle 2018, 17, 829–832. [Google Scholar] [CrossRef]
- Mou, Y.; Zhang, Y.; Wu, J.; Hu, B.; Zhang, C.; Duan, C.; Li, B. The landscape of iron metabolism-related and methylated genes in the prognosis prediction of clear cell renal cell carcinoma. Front. Oncol. 2020, 10, 788. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamenkovic, A.; Pierce, G.N.; Ravandi, A. Phospholipid oxidation products in ferroptotic myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H156–H163. [Google Scholar] [CrossRef]
- Jiang, M.; Qiao, M.; Zhao, C.; Deng, J.; Li, X.; Zhou, C. Targeting ferroptosis for cancer therapy: Exploring novel strategies from its mechanisms and role in cancers. Transl. Lung Cancer Res. 2020, 9, 1569. [Google Scholar] [CrossRef]
- Lu, J.; Yang, J.; Zheng, Y.; Chen, X.; Fang, S. Extracellular vesicles from endothelial progenitor cells prevent steroid-induced osteoporosis by suppressing the ferroptotic pathway in mouse osteoblasts based on bioinformatics evidence. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F.; Mansoori, O. Effect of valproic acid in comparison with vorinostat on cell growth inhibition and apoptosis induction in the human colon cancer SW48 cells in vitro. Exp. Oncol. 2018. Available online: http://dspace.nbuv.gov.ua/handle/123456789/145572 (accessed on 10 September 2021).
- Miyamoto, K.; Watanabe, M.; Boku, S.; Sukeno, M.; Morita, M.; Kondo, H.; Sakaguchi, K.; Taguchi, T.; Sakai, T. xCT inhibition increases sensitivity to vorinostat in a ROS-dependent manner. Cancers 2020, 12, 827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhao, L.; Gao, Y.; Yao, F.; Marti, T.M.; Schmid, R.A.; Peng, R.-W. Pharmacotranscriptomic analysis reveals novel drugs and gene networks regulating ferroptosis in cancer. Cancers 2020, 12, 3273. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential applications of NRF2 inhibitors in cancer therapy. Oxidative Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Sato, E.; Ito, J.; Yamada, K.-i.; Suzuki, C.; Oikawa, Y.; Matsuhashi, T.; Kikuchi, K.; Toyohara, T.; Suzuki, T. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J. Am. Soc. Nephrol. 2020, 31, 280–296. [Google Scholar] [CrossRef]
- Pan, Y.; Tang, P.; Cao, J.; Song, Q.; Zhu, L.; Ma, S.; Zhang, J. Lipid peroxidation aggravates anti-tuberculosis drug-induced liver injury: Evidence of ferroptosis induction. Biochem. Biophys. Res. Commun. 2020, 533, 1512–1518. [Google Scholar] [CrossRef]
- Tsvetkova, D.; Obreshkova, D.; Zheleva-Dimitrova, D.; Saso, L. Antioxidant activity of galantamine and some of its derivatives. Curr. Med. Chem. 2013, 20, 4595–4608. [Google Scholar] [CrossRef]
- Nie, F.; Liang, Y.; Jiang, B.; Li, X.; Xun, H.; He, W.; Lau, H.T.; Ma, X. Apoptotic effect of tannic acid on fatty acid synthase over-expressed human breast cancer cells. Tumor Biol. 2016, 37, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, S.; Fu, Q.; Smith, K.; Tu, K.; Li, H.; Zhao, Y. FASN, ErbB2-mediated glycolysis is required for breast cancer cell migration. Oncol. Rep. 2016, 35, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhou, L.; Li, H.; Tian, Y.; Li, J.; Dong, L.; Zhao, Y.; Wei, D. Fatty acid synthase affects expression of ErbB receptors in epithelial to mesenchymal transition of breast cancer cells and invasive ductal carcinoma. Oncol. Lett. 2017, 14, 5934–5946. [Google Scholar] [CrossRef] [Green Version]
- Farhat, D.; Ghayad, S.E.; Icard, P.; Le Romancer, M.; Hussein, N.; Lincet, H. Lipoic acid-induced oxidative stress abrogates IGF-1R maturation by inhibiting the CREB/furin axis in breast cancer cell lines. Oncogene 2020, 39, 3604–3610. [Google Scholar] [CrossRef]
- Farhat, D.; Léon, S.; Ghayad, S.E.; Gadot, N.; Icard, P.; Le Romancer, M.; Hussein, N.; Lincet, H. Lipoic acid decreases breast cancer cell proliferation by inhibiting IGF-1R via furin downregulation. Br. J. Cancer 2020, 122, 885–894. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, J.H.; Jang, S.J.; Yun, J.W.; Kang, K.M.; Jeong, H.; Ha, I.B.; Jeong, B.K. Synergistic tumoricidal effects of alpha-lipoic acid and radiotherapy on human breast cancer cells via HMGB1. Cancer Res. Treat. 2020, 53, 685. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Durán, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Long, J.-P.; Li, X.-N.; Zhang, F. Targeting metabolism in breast cancer: How far we can go? World J. Clin. Oncol. 2016, 7, 122. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Wang, X. The metabolic mechanisms of breast cancer metastasis. Front. Oncol. 2021, 10, 2942. [Google Scholar] [CrossRef]
- Gong, Y.; Ji, P.; Yang, Y.-S.; Xie, S.; Yu, T.-J.; Xiao, Y.; Jin, M.-L.; Ma, D.; Guo, L.-W.; Pei, Y.-C. Metabolic-pathway-based subtyping of triple-negative breast cancer reveals potential therapeutic targets. Cell Metab. 2021, 33, 51–64. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 1–14. [Google Scholar] [CrossRef]
- Bevinakoppamath, S.; Ahmed, A.M.S.; Ramachandra, S.C.; Vishwanath, P.; Prashant, A. Chemopreventive and anticancer property of selenoproteins in obese breast cancer. Front. Pharmacol. 2021, 12, 618172. [Google Scholar] [CrossRef]
- Wang, H.; You, S.; Fang, M.; Fang, Q. Recognition of immune microenvironment landscape and immune-related prognostic genes in breast cancer. BioMed Res. Int. 2020, 2020, 3909416. [Google Scholar] [CrossRef]
- Sousa, B.; Pereira, J.; Marques, R.; Grilo, L.F.; Pereira, S.P.; Sardão, V.A.; Schmitt, F.; Oliveira, P.J.; Paredes, J. P-cadherin induces anoikis-resistance of matrix-detached breast cancer cells by promoting pentose phosphate pathway and decreasing oxidative stress. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165964. [Google Scholar] [CrossRef] [PubMed]
- Schömel, N.; Gruber, L.; Alexopoulos, S.J.; Trautmann, S.; Olzomer, E.M.; Byrne, F.L.; Hoehn, K.L.; Gurke, R.; Thomas, D.; Ferreirós, N. UGCG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Sci. Rep. 2020, 10, 8182. [Google Scholar] [CrossRef]
- Helm, J.S.; Rudel, R.A. Adverse outcome pathways for ionizing radiation and breast cancer involve direct and indirect DNA damage, oxidative stress, inflammation, genomic instability, and interaction with hormonal regulation of the breast. Arch. Toxicol. 2020, 94, 1511–1549. [Google Scholar] [CrossRef]
- Elias, A.D. Triple-negative breast cancer: A short review. Am. J. Clin. Oncol. 2010, 33, 637–645. [Google Scholar] [CrossRef]
- Matsuda, N.; Lim, B.; Wang, X.; Ueno, N.T. Early clinical development of epidermal growth factor receptor targeted therapy in breast cancer. Expert Opin. Investig. Drugs 2017, 26, 463–479. [Google Scholar] [CrossRef]
- Weng, T.-H.; Yao, M.-Y.; Xu, X.-M.; Hu, C.-Y.; Yao, S.-H.; Liu, Y.-Z.; Wu, Z.-G.; Tang, T.-M.; Fu, P.-F.; Wang, M.-H. RON and MET co-overexpression are significant pathological characteristics of poor survival and therapeutic targets of tyrosine kinase inhibitors in triple-negative breast cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2020, 52, 973. [Google Scholar] [CrossRef]
- Xiang, H.; Zhang, J.; Lin, C.; Zhang, L.; Liu, B.; Ouyang, L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm. Sin. B 2020, 10, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Gay, L.M. Comprehensive genomic sequencing and the molecular profiles of clinically advanced breast cancer. Pathology 2017, 49, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Nowacka-Zawisza, M.; Krajewska, W.M. Triple-negative breast cancer: Molecular characteristics and potential therapeutic approaches. Postepy Hig. I Med. Dosw. 2013, 67, 1090–1097. [Google Scholar] [CrossRef]
- Vijay, S.; Gujral, T.S. Non-linear deep neural network for rapid and accurate prediction of phenotypic responses to kinase inhibitors. Iscience 2020, 23, 101129. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpi, A.; Nikolettos, K.; Vavourakis, M.; Diamantis, E.; Patsouras, A.; Farmaki, P.; Nonni, A.; Dimitroulis, D.; Mantas, D. Triple-negative breast cancer: The progress of targeted therapies and future tendencies. Anticancer. Res. 2019, 39, 5285–5296. [Google Scholar] [CrossRef]
- Tolba, M.F.; Elghazaly, H.; Bousoik, E.; Elmazar, M.M.A.; Tolaney, S.M. Novel combinatorial strategies for boosting the efficacy of immune checkpoint inhibitors in advanced breast cancers. Clin. Transl. Oncol. 2021, 23, 1979–1994. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zhou, Q.; Zhang, Y.; Zhou, W.; Wu, Y.; Zhen, C.; Zhang, M.; Fan, F.; Chen, G.; Zhao, J. EGFR tyrosine kinase inhibitor HS-10296 induces autophagy and apoptosis in triplenegative breast cancer MDA-MB-231 cells. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2020, 40, 981–987. [Google Scholar]
- Kawai, M.; Nakashima, A.; Kamada, S.; Kikkawa, U. Midostaurin preferentially attenuates proliferation of triple-negative breast cancer cell lines through inhibition of Aurora kinase family. J. Biomed. Sci. 2015, 22, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, K.S.; Yi, Y.W.; Cho, J.; Seong, Y.-S. Dual inhibition of AKT and MEK pathways potentiates the anti-cancer effect of gefitinib in triple-negative breast cancer cells. Cancers 2021, 13, 1205. [Google Scholar] [CrossRef]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular pathways: Receptor ectodomain shedding in treatment, resistance, and monitoring of cancer. Clin. Cancer Res. 2017, 23, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, Y.; Higashisaka, K.; Yang, L.; Sekine, N.; Lin, Y.; Tsujino, H.; Nagano, K.; Tsutsumi, Y. Inhibition of Akt/mTOR pathway overcomes intrinsic resistance to dasatinib in triple-negative breast cancer. Biochem. Biophys. Res. Commun. 2020, 533, 672–678. [Google Scholar] [CrossRef]
- Lux, M.P.; Schneeweiss, A.; Hartkopf, A.D.; Müller, V.; Janni, W.; Belleville, E.; Stickeler, E.; Thill, M.; Fasching, P.A.; Kolberg, H.-C. Update breast cancer 2020 part 5–moving therapies from advanced to early breast cancer patients. Geburtshilfe Und Frauenheilkd. 2021, 81, 469–480. [Google Scholar] [CrossRef]
- Malhotra, M.K.; Emens, L.A. The evolving management of metastatic triple negative breast cancer. Semin. Oncol. 2020, 47, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Vinik, Y.; Saroha, A.; Nair, N.U.; Ruppin, E.; Mills, G.; Karn, T.; Dubey, V.; Khera, L.; Raj, H.; et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 2020, 6, eaba8968. [Google Scholar] [CrossRef] [PubMed]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Göbl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y. Possible beneficial effects of n-acetylcysteine for treatment of triple-negative breast cancer. Antioxidants 2021, 10, 169. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Taie, Z.; Hannink, M.; Mitchem, J.; Papageorgiou, C.; Shyu, C.-R. Drug Repositioning and Subgroup Discovery for Precision Medicine Implementation in Triple Negative Breast Cancer. Cancers 2021, 13, 6278. https://doi.org/10.3390/cancers13246278
Al-Taie Z, Hannink M, Mitchem J, Papageorgiou C, Shyu C-R. Drug Repositioning and Subgroup Discovery for Precision Medicine Implementation in Triple Negative Breast Cancer. Cancers. 2021; 13(24):6278. https://doi.org/10.3390/cancers13246278
Chicago/Turabian StyleAl-Taie, Zainab, Mark Hannink, Jonathan Mitchem, Christos Papageorgiou, and Chi-Ren Shyu. 2021. "Drug Repositioning and Subgroup Discovery for Precision Medicine Implementation in Triple Negative Breast Cancer" Cancers 13, no. 24: 6278. https://doi.org/10.3390/cancers13246278
APA StyleAl-Taie, Z., Hannink, M., Mitchem, J., Papageorgiou, C., & Shyu, C.-R. (2021). Drug Repositioning and Subgroup Discovery for Precision Medicine Implementation in Triple Negative Breast Cancer. Cancers, 13(24), 6278. https://doi.org/10.3390/cancers13246278