
Anti-PD-1 Therapy with Adjuvant Ablative Fractional Laser Improves Anti-Tumor Response in Basal Cell Carcinomas

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
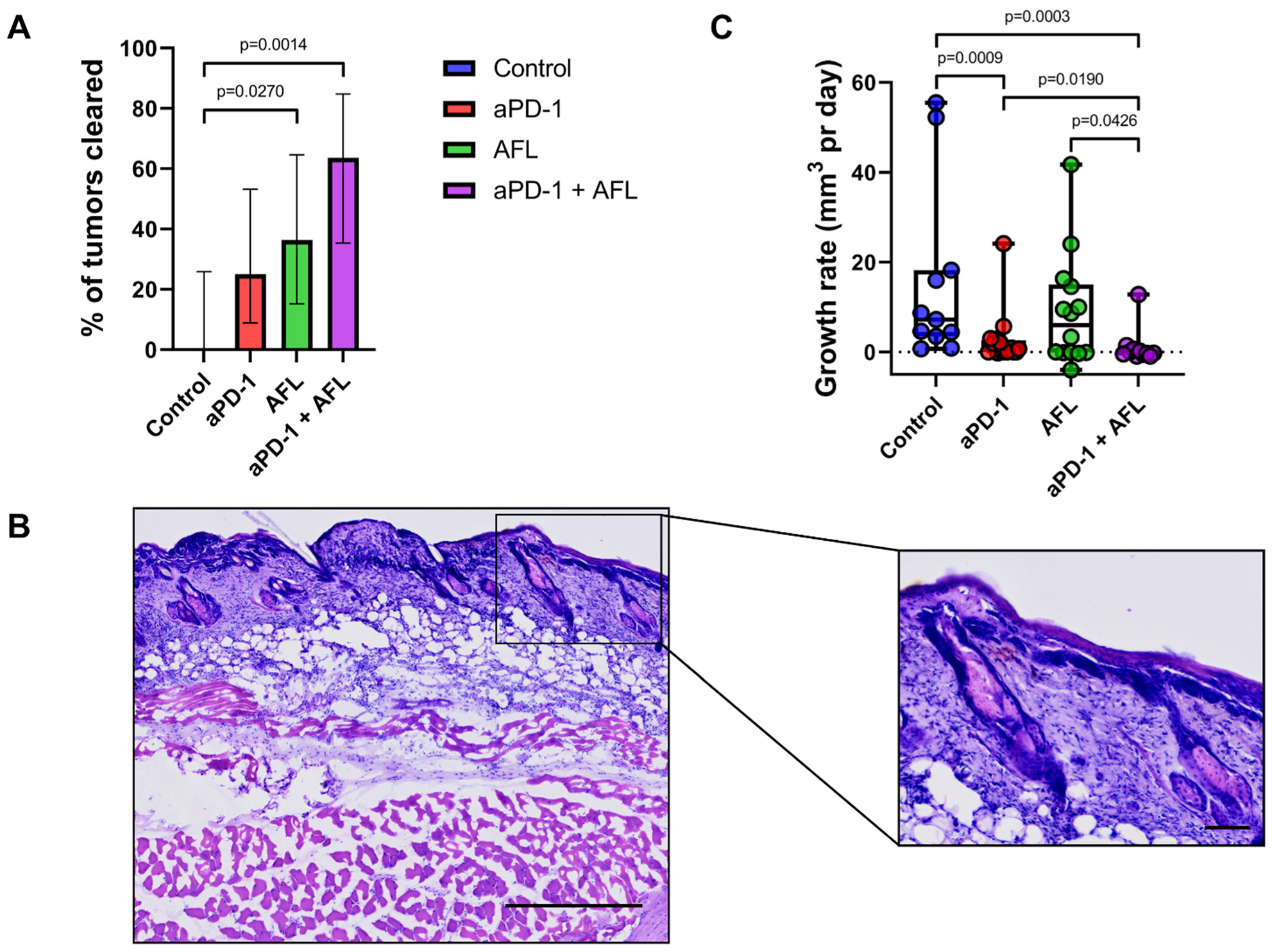
2.1. Tumor Response
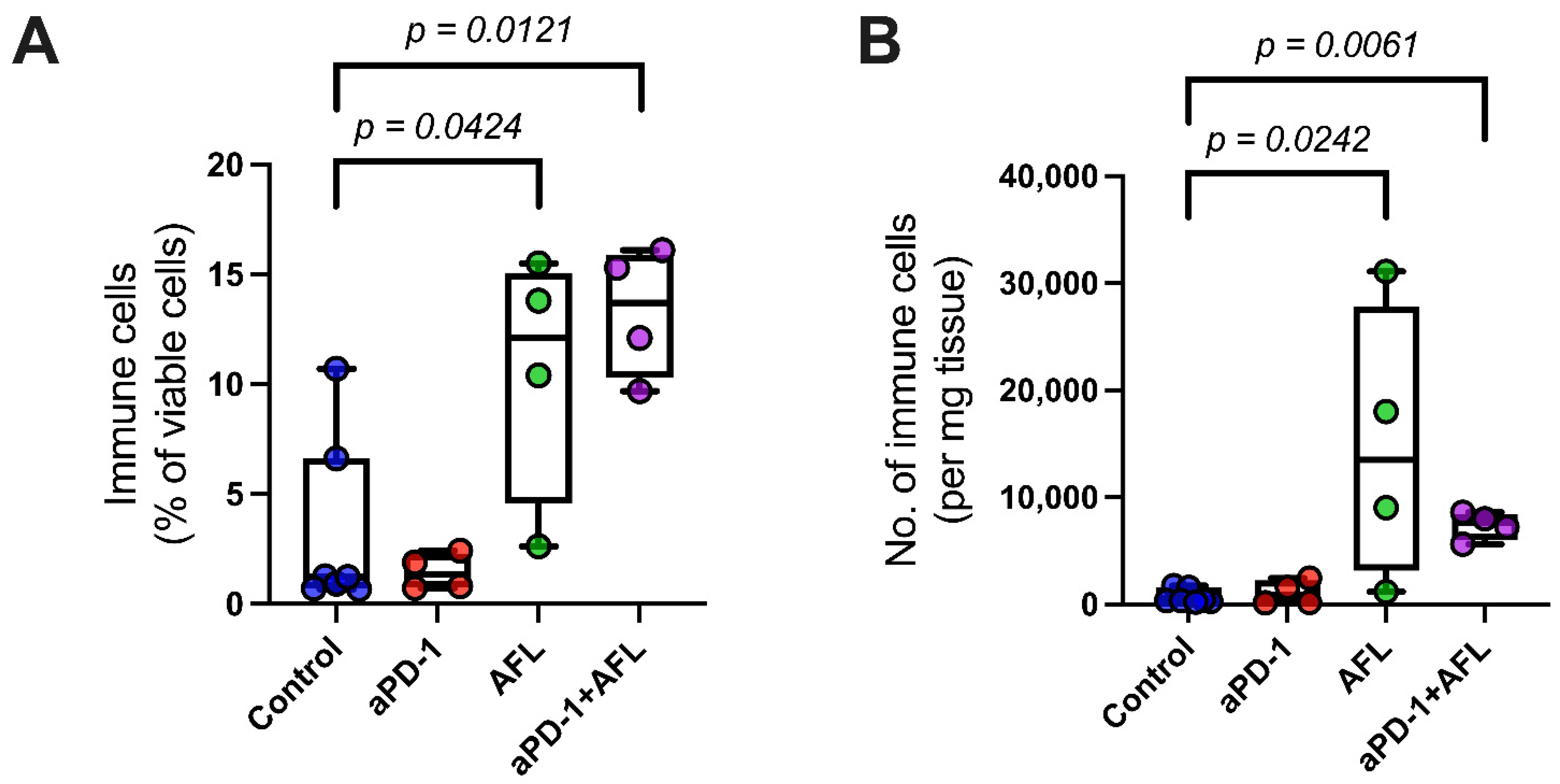
2.2. Tumor Immune Infiltration
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Study Design
4.3. Treatment with aPD-1 and AFL
4.4. Tumor Response
4.5. Flow Cytometry
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Didona, D.; Morese, R.; Campana, I.; Coco, V.; Di Pietro, F.R.; Ricci, F.; Pallotta, S.; Candi, E.; Abeni, D.; et al. Basal Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2020, 8, 449. [Google Scholar] [CrossRef] [PubMed]
- Madani, S.; Marwaha, S.; Dusendang, J.R.; Alexeeff, S.; Pham, N.; Chen, E.H.; Han, S.; Herrinton, L.J. Ten-Year Follow-up of Persons With Sun-Damaged Skin Associated With Subsequent Development of Cutaneous Squamous Cell Carcinoma. JAMA Dermatol. 2021, 157, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Conforti, C.; Corneli, P.; Harwood, C.; Zalaudek, I. Evolving Role of Systemic Therapies in Non-melanoma Skin Cancer. Clin. Oncol. 2019, 31, 759–768. [Google Scholar] [CrossRef]
- Ahluwalia, J.; Avram, M.M.; Ortiz, A.E. The Evolving Story of Laser Therapeutics for Basal Cell Carcinoma. Dermatol. Surg. 2020, 46, 1045–1053. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- van den Bulk, J.; Verdegaal, E.M.; de Miranda, N.F. Cancer immunotherapy: Broadening the scope of targetable tumours. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Erlendsson, A.M.; Olesen, U.H.; Haedersdal, M.; Rossi, A.M. Ablative fractional laser-assisted treatments for keratinocyte carcinomas and its precursors-Clinical review and future perspectives. Adv. Drug Deliv. Rev. 2020, 153, 185–194. [Google Scholar] [CrossRef]
- Markham, A.; Duggan, S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Hwang, S.J.E.; Anforth, R.; Carlos, G.; Chou, S.; Carlino, M.; Fernandez-Penas, P. Incidence of Basal Cell Carcinoma and Squamous Cell Carcinoma in Patients on Antiprogrammed Cell Death-1 Therapy for Metastatic Melanoma. J. Immunother. 2018, 41, 343–349. [Google Scholar] [CrossRef]
- Falchook, G.S.; Leidner, R.; Stankevich, E.; Piening, B.; Bifulco, C.; Lowy, I.; Fury, M.G. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J. Immunother. Cancer 2016, 4, 70. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Goodman, A.M.; Cohen, P.R.; Jensen, T.J.; Ellison, C.K.; Frampton, G.; Miller, V.; Patel, S.P.; Kurzrock, R. Metastatic basal cell carcinoma with amplification of PD-L1: Exceptional response to anti-PD1 therapy. NPJ Genom. Med. 2016, 1, 16037. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.R.; Kato, S.; Goodman, A.M.; Ikeda, S.; Kurzrock, R. Appearance of New Cutaneous Superficial Basal Cell Carcinomas during Successful Nivolumab Treatment of Refractory Metastatic Disease: Implications for Immunotherapy in Early Versus Late Disease. Int. J. Mol. Sci. 2017, 18, 1663. [Google Scholar] [CrossRef]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Prey, S.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: An open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 848–857. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Gao, L.; Liu, Y.; Meng, F.; Li, X.; Qin, F.X. Targeting the tumor microenvironment to overcome immune checkpoint blockade therapy resistance. Immunol. Lett. 2020, 220, 88–96. [Google Scholar] [CrossRef]
- Olesen, U.H.; Clergeaud, G.; Hendel, K.K.; Yeung, K.; Lerche, C.M.; Andresen, T.L.; Haedersdal, M. Enhanced and Sustained Cutaneous Delivery of Vismodegib by Ablative Fractional Laser and Microemulsion Formulation. J. Investig. Dermatol. 2020, 140, 2051–2059. [Google Scholar] [CrossRef]
- Kawakubo, M.; Cunningham, T.J.; Demehri, S.; Manstein, D. Fractional Laser Releases Tumor-Associated Antigens in Poorly Immunogenic Tumor and Induces Systemic Immunity. Sci. Rep. 2017, 7, 12751. [Google Scholar] [CrossRef] [Green Version]
- Kawakubo, M.; Demehri, S.; Manstein, D. Fractional laser exposure induces neutrophil infiltration (N1 phenotype) into the tumor and stimulates systemic anti-tumor immune response. PLoS ONE 2017, 12, e0184852. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Shi, L.; Zhang, F.; Zhou, F.; Zhang, L.; Wang, B.; Wang, P.; Zhang, Y.; Zhang, H.; Yang, D.; et al. Laser immunotherapy for cutaneous squamous cell carcinoma with optimal thermal effects to enhance tumour immunogenicity. Int. J. Hyperth. 2018, 34, 1337–1350. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, J.; Zhang, Y.; Liu, M.; Lang, M.L.; Li, M.; Chen, W.R. Local Phototherapy Synergizes with Immunoadjuvant for Treatment of Pancreatic Cancer through Induced Immunogenic Tumor Vaccine. Clin. Cancer Res. 2018, 24, 5335–5346. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.A.; Kawakubo, M.; Juneja, V.R.; Su, M.Y.; Erlich, T.H.; LaFleur, M.W.; Kemeny, L.V.; Rashid, M.; Malehmir, M.; Rabi, S.A.; et al. Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Omland, S.H.; Wenande, E.C.; Svane, I.M.; Tam, J.; Olesen, U.H.; Haedersdal, M. Laser Immunotherapy: A Potential Treatment Modality for Keratinocyte Carcinoma. Cancers 2021, 13, 5405. [Google Scholar] [CrossRef]
- Prignano, F.; Campolmi, P.; Bonan, P.; Ricceri, F.; Cannarozzo, G.; Troiano, M.; Lotti, T. Fractional CO2 laser: A novel therapeutic device upon photobiomodulation of tissue remodeling and cytokine pathway of tissue repair. Dermatol. Ther. 2009, 22, S8–S15. [Google Scholar] [CrossRef]
- Helbig, D.; Bodendorf, M.O.; Grunewald, S.; Kendler, M.; Simon, J.C.; Paasch, U. Immunohistochemical investigation of wound healing in response to fractional photothermolysis. J. Biomed. Opt. 2009, 14, 064044. [Google Scholar] [CrossRef] [Green Version]
- Fontenete, S.; Lerche, C.M.; Paasch, U.; Perez-Moreno, M.; Olesen, U.H.; Haedersdal, M. Tumor Clearance and Immune Cell Recruitment in UV-Induced Murine Squamous Cell Carcinoma Exposed to Ablative Fractional Laser and Imiquimod Treatment. Lasers Surg. Med. 2021, 53, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.; Wang, G.; Dolorito, J.A.; Kc, S.; Libove, E.; Epstein, E.H., Jr. Vitamin D3 Produced by Skin Exposure to UVR Inhibits Murine Basal Cell Carcinoma Carcinogenesis. J. Investig. Dermatol. 2017, 137, 2613–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.C.; Loewer, M.; Boegel, S.; de Graaf, J.; Bender, C.; Tadmor, A.D.; Boisguerin, V.; Bukur, T.; Sorn, P.; Paret, C.; et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genom. 2014, 15, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.A.; Hughes, A.M.; Walton, J.; Coenen-Stass, A.M.L.; Magiera, L.; Mooney, L.; Bell, S.; Staniszewska, A.D.; Sandin, L.C.; Barry, S.T.; et al. Longitudinal immune characterization of syngeneic tumor models to enable model selection for immune oncology drug discovery. J. Immunother. Cancer 2019, 7, 328. [Google Scholar] [CrossRef]
- Jensen, H.K.; Donskov, F.; Marcussen, N.; Nordsmark, M.; Lundbeck, F.; von der Maase, H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J. Clin. Oncol. 2009, 27, 4709–4717. [Google Scholar] [CrossRef]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98259. [Google Scholar] [CrossRef] [Green Version]
- Berry, R.S.; Xiong, M.J.; Greenbaum, A.; Mortaji, P.; Nofchissey, R.A.; Schultz, F.; Martinez, C.; Luo, L.; Morris, K.T.; Hanson, J.A. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PLoS ONE 2017, 12, e0188799. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Alatrash, G.; Qiao, N.; Wu, Y.; Sukhumalchandra, P.; St John, L.S.; Philips, A.V.; Xiao, H.; Zhang, M.; Ruisaard, K.; et al. Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res. 2012, 72, 3153–3162. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Qiu, L.; Li, Z.; Wang, X.Y.; Yi, H. Understanding the Multifaceted Role of Neutrophils in Cancer and Autoimmune Diseases. Front. Immunol. 2018, 9, 2456. [Google Scholar] [CrossRef]
- Wenande, E.; Tam, J.; Bhayana, B.; Schlosser, S.K.; Ishak, E.; Farinelli, W.A.; Chlopik, A.; Hoang, M.P.; Pinkhasov, O.R.; Caravan, P.; et al. Laser-assisted delivery of synergistic combination chemotherapy in in vivo skin. J. Control. Release 2018, 275, 242–253. [Google Scholar] [CrossRef]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Lore, K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Culshaw, S.; Millington, O.R.; Brewer, J.M.; McInnes, I.B. Murine neutrophils present Class II restricted antigen. Immunol. Lett. 2008, 118, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, H.; Geng, S.; Lu, R.; Okamoto, T.; Yao, Y.; Mayuzumi, N.; Kotol, P.F.; Chojnacki, B.J.; Miyazaki, T.; Gallo, R.L.; et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood 2013, 121, 1677–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radsak, M.; Iking-Konert, C.; Stegmaier, S.; Andrassy, K.; Hansch, G.M. Polymorphonuclear neutrophils as accessory cells for T-cell activation: Major histocompatibility complex class II restricted antigen-dependent induction of T-cell proliferation. Immunology 2000, 101, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Wang, J.; Mancianti, M.L.; Epstein, E.H., Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Zhu, G.A.; Cheung, C.; Li, S.; Kim, J.; Chang, A.L. Association Between Programmed Death Ligand 1 Expression in Patients With Basal Cell Carcinomas and the Number of Treatment Modalities. JAMA Dermatol. 2017, 153, 285–290. [Google Scholar] [CrossRef]
- Gompertz-Mattar, M.; Perales, J.; Sahu, A.; Mondaca, S.; Gonzalez, S.; Uribe, P.; Navarrete-Dechent, C. Differential expression of programmed cell death ligand 1 (PD-L1) and inflammatory cells in basal cell carcinoma subtypes. Arch. Dermatol. Res. 2021. [Google Scholar] [CrossRef]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Nitzki, F.; Becker, M.; Frommhold, A.; Schulz-Schaeffer, W.; Hahn, H. Patched knockout mouse models of Basal cell carcinoma. J. Skin Cancer 2012, 2012, 907543. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Tang, J.Y.; Li, D.; Reich, M.; Callahan, C.A.; Fu, L.; Yauch, R.L.; Wang, F.; Kotkow, K.; Chang, K.S.; et al. Targeting superficial or nodular Basal cell carcinoma with topically formulated small molecule inhibitor of smoothened. Clin. Cancer Res. 2011, 17, 3378–3387. [Google Scholar] [CrossRef] [Green Version]
- Aszterbaum, M.; Epstein, J.; Oro, A.; Douglas, V.; LeBoit, P.E.; Scott, M.P.; Epstein, E.H., Jr. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat. Med. 1999, 5, 1285–1291. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olesen, U.H.; Wiinberg, M.; Lerche, C.M.; Jæhger, D.E.; Andresen, T.L.; Haedersdal, M. Anti-PD-1 Therapy with Adjuvant Ablative Fractional Laser Improves Anti-Tumor Response in Basal Cell Carcinomas. Cancers 2021, 13, 6326. https://doi.org/10.3390/cancers13246326
Olesen UH, Wiinberg M, Lerche CM, Jæhger DE, Andresen TL, Haedersdal M. Anti-PD-1 Therapy with Adjuvant Ablative Fractional Laser Improves Anti-Tumor Response in Basal Cell Carcinomas. Cancers. 2021; 13(24):6326. https://doi.org/10.3390/cancers13246326
Chicago/Turabian StyleOlesen, Uffe Høgh, Martin Wiinberg, Catharina Margrethe Lerche, Ditte Elisabeth Jæhger, Thomas Lars Andresen, and Merete Haedersdal. 2021. "Anti-PD-1 Therapy with Adjuvant Ablative Fractional Laser Improves Anti-Tumor Response in Basal Cell Carcinomas" Cancers 13, no. 24: 6326. https://doi.org/10.3390/cancers13246326
APA StyleOlesen, U. H., Wiinberg, M., Lerche, C. M., Jæhger, D. E., Andresen, T. L., & Haedersdal, M. (2021). Anti-PD-1 Therapy with Adjuvant Ablative Fractional Laser Improves Anti-Tumor Response in Basal Cell Carcinomas. Cancers, 13(24), 6326. https://doi.org/10.3390/cancers13246326