
A Multidisciplinary Diagnostic Approach Reveals a Higher Prevalence of Indolent Systemic Mastocytosis: 15-Years’ Experience of the GISM Network

 ,
,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Reason for Referral
3.2. Final Diagnosis
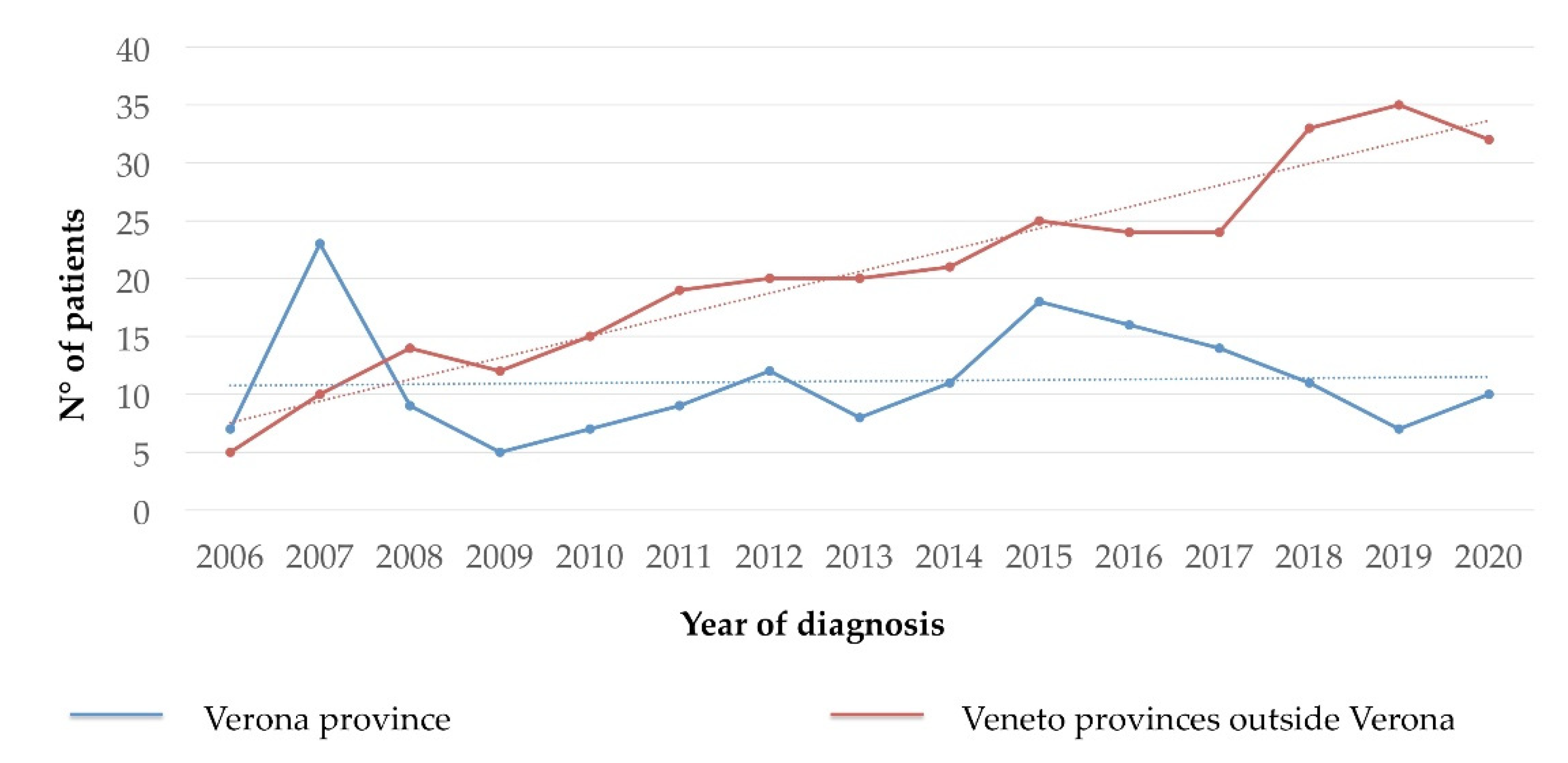
3.3. Prevalence and Incidence of CMD
3.4. Clinical, Laboratory, and Diagnostic Characteristics of Patients
3.5. Hymenoptera Venom Allergy and Other Allergic Reactions
3.6. Bone Involvement
3.7. Treatment of Advanced SM
3.8. Survival
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, H.C.; Hamdy, N.A.T.; et al. Standards and Standardization in Mastocytosis: Consensus Statements on Diagnostics, Treatment Recommendations and Response Criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef]
- Horny, H.P.; Akin, C.; Arber, D.; Peterson, L.A.; Tefferi, A.; Metcalfe, D.D.; Bennett, J.M.; Bain, B.; Escribano, L.; Valent, P. Mastocytosis. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC Press: Lyon, France, 2017; pp. 62–69. [Google Scholar]
- Sonneck, K.; Florian, S.; Müllauer, L.; Wimazal, F.; Födinger, M.; Sperr, W.R.; Valent, P. Diagnostic and Subdiagnostic Accumulation of Mast Cells in the Bone Marrow of Patients with Anaphylaxis: Monoclonal Mast Cell Activation Syndrome. Int. Arch. Allergy Immunol. 2007, 142, 158–164. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Arock, M.; Brockow, K.; Butterfield, J.H.; Carter, M.C.; Castells, M.; Escribano, L.; Hartmann, K.; Lieberman, P.; et al. Definitions, Criteria and Global Classification of Mast Cell Disorders with Special Reference to Mast Cell Activation Syndromes: A Consensus Proposal. Int. Arch. Allergy Immunol. 2012, 157, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Bonadonna, P.; Perbellini, O.; Passalacqua, G.; Caruso, B.; Colarossi, S.; Dal Fior, D.; Castellani, L.; Bonetto, C.; Frattini, F.; Dama, A.; et al. Clonal Mast Cell Disorders in Patients with Systemic Reactions to Hymenoptera Stings and Increased Serum Tryptase Levels. J. Allergy Clin. Immunol. 2009, 123, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Gülen, T.; Hägglund, H.; Sander, B.; Dahlén, B.; Nilsson, G. The Presence of Mast Cell Clonality in Patients with Unexplained Anaphylaxis. Clin. Exp. Allergy 2014, 44, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, R.; Bonadonna, P.; Bonifacio, M.; Artuso, A.; Schena, D.; Rossini, M.; Perbellini, O.; Colarossi, S.; Chilosi, M.; Pizzolo, G. Isolated Bone Marrow Mastocytosis: An Underestimated Subvariant of Indolent Systemic Mastocytosis. Haematologica 2011, 96, 482–484. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, R.; Bonifacio, M.; Lucchini, G.; Sperr, W.R.; Scaffidi, L.; van Anrooij, B.; Elberink, H.N.O.; Rossignol, J.; Hermine, O.; Gorska, A.; et al. Refined Diagnostic Criteria for Bone Marrow Mastocytosis: A Proposal of the European Competence Network on Mastocytosis. Leukemia 2021. e-pub ahead of print. [Google Scholar] [CrossRef]
- Brockow, K. Epidemiology, Prognosis, and Risk Factors in Mastocytosis. Immunol. Allergy Clin. 2014, 34, 283–295. [Google Scholar] [CrossRef]
- Lim, K.-H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.-Y.; Pardanani, A. Systemic Mastocytosis in 342 Consecutive Adults: Survival Studies and Prognostic Factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.S.; Skovbo, S.; Vestergaard, H.; Kristensen, T.; Møller, M.; Bindslev-Jensen, C.; Fryzek, J.P.; Broesby-Olsen, S. Epidemiology of Systemic Mastocytosis in Denmark. Br. J. Haematol. 2014, 166, 521–528. [Google Scholar] [CrossRef]
- van Doormaal, J.J.; Arends, S.; Brunekreeft, K.L.; van der Wal, V.B.; Sietsma, J.; van Voorst Vader, P.C.; Oude Elberink, J.N.G.; Kluin-Nelemans, J.C.; van der Veer, E.; de Monchy, J.G.R. Prevalence of Indolent Systemic Mastocytosis in a Dutch Region. J. Allergy Clin. Immunol. 2013, 131, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Oude Elberink, J.N.G.; Gorska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International Prognostic Scoring System for Mastocytosis (IPSM): A Retrospective Cohort Study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef]
- Tanasi, I.; Bonifacio, M.; Pizzolato, M.; Irene Grifoni, F.; Sciumè, M.; Elena, C.; Benvenuti, P.; Mannelli, F.; Parente, R.; Schena, D.; et al. Familial Occurrence of Systemic and Cutaneous Mastocytosis in an Adult Multicentre Series. Br. J. Haematol. 2021, 193, 845–848. [Google Scholar] [CrossRef]
- Orsolini, G.; Viapiana, O.; Rossini, M.; Bonifacio, M.; Zanotti, R. Bone Disease in Mastocytosis. Immunol. Allergy Clin. 2018, 38, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Zanotti, R.; Orsolini, G.; Tripi, G.; Viapiana, O.; Idolazzi, L.; Zamò, A.; Bonadonna, P.; Kunnathully, V.; Adami, S.; et al. Prevalence, Pathogenesis, and Treatment Options for Mastocytosis-Related Osteoporosis. Osteoporos. Int. 2016, 27, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Oude Elberink, J.N.; de Monchy, J.G.; Kors, J.W.; van Doormaal, J.J.; Dubois, A.E. Fatal Anaphylaxis after a Yellow Jacket Sting, despite Venom Immunotherapy, in Two Patients with Mastocytosis. J. Allergy Clin. Immunol. 1997, 99, 153–154. [Google Scholar] [CrossRef]
- Bonadonna, P.; Bonifacio, M.; Lombardo, C.; Zanotti, R. Hymenoptera Allergy and Mast Cell Activation Syndromes. Curr. Allergy Asthma Rep. 2016, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Perbellini, O.; Zamò, A.; Colarossi, S.; Zampieri, F.; Zoppi, F.; Bonadonna, P.; Schena, D.; Artuso, A.; Martinelli, G.; Chilosi, M.; et al. Primary Role of Multiparametric Flow Cytometry in the Diagnostic Work-up of Indolent Clonal Mast Cell Disorders. Cytom. B Clin. Cytom. 2011, 80, 362–368. [Google Scholar] [CrossRef]
- De Matteis, G.; Zanotti, R.; Colarossi, S.; De Benedittis, C.; Garcia-Montero, A.; Bonifacio, M.; Sartori, M.; Aprili, F.; Caruso, B.; Paviati, E.; et al. The Impact of Sensitive KIT D816V Detection on Recognition of Indolent Systemic Mastocytosis. Leuk. Res. 2015, 39, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Kanis, J.A. Assessment of Fracture Risk and Its Application to Screening for Postmenopausal Osteoporosis: Synopsis of a WHO Report. WHO Study Group. Osteoporos. Int. 1994, 4, 368–381. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, L.; Alvarez-Twose, I.; García-Montero, A.C.; Teodosio, C.; Jara-Acevedo, M.; Pedreira, C.E.; Matito, A.; Morgado, J.M.T.; Sánchez, M.L.; Mollejo, M.; et al. Evaluation of the WHO Criteria for the Classification of Patients with Mastocytosis. Mod. Pathol. 2011, 24, 1157–1168. [Google Scholar] [CrossRef] [Green Version]
- Wimazal, F.; Geissler, P.; Shnawa, P.; Sperr, W.R.; Valent, P. Severe Life-Threatening or Disabling Anaphylaxis in Patients with Systemic Mastocytosis: A Single-Center Experience. Int. Arch. Allergy Immunol 2012, 157, 399–405. [Google Scholar] [CrossRef]
- Pieri, L.; Bonadonna, P.; Elena, C.; Papayannidis, C.; Grifoni, F.I.; Rondoni, M.; Girlanda, S.; Mauro, M.; Magliacane, D.; Elli, E.M.; et al. Clinical Presentation and Management Practice of Systemic Mastocytosis. A Survey on 460 Italian Patients. Am. J. Hematol. 2016, 91, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Gotlib, J.; Gerds, A.T.; Bose, P.; Castells, M.C.; Deininger, M.W.; Gojo, I.; Gundabolu, K.; Hobbs, G.; Jamieson, C.; McMahon, B.; et al. Systemic Mastocytosis, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1500–1537. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Twose, I.; González de Olano, D.; Sánchez-Muñoz, L.; Matito, A.; Esteban-López, M.I.; Vega, A.; Mateo, M.B.; Alonso Díaz de Durana, M.D.; de la Hoz, B.; Del Pozo Gil, M.D.; et al. Clinical, Biological, and Molecular Characteristics of Clonal Mast Cell Disorders Presenting with Systemic Mast Cell Activation Symptoms. J. Allergy Clin. Immunol. 2010, 125, 1269–1278. [Google Scholar] [CrossRef]
- Kluin-Nelemans, H.C.; Jawhar, M.; Reiter, A.; van Anrooij, B.; Gotlib, J.; Hartmann, K.; Illerhaus, A.; Elberink, H.N.G.O.; Gorska, A.; Niedoszytko, M.; et al. Cytogenetic and Molecular Aberrations and Worse Outcome for Male Patients in Systemic Mastocytosis. Theranostics 2020, 11, 292–303. [Google Scholar] [CrossRef]
- Schwaab, J.; O Hartmann, N.C.D.; Naumann, N.; Jawhar, M.; Weiß, C.; Metzgeroth, G.; Schmid, A.; Lübke, J.; Reiter, L.; Fabarius, A.; et al. Importance of Adequate Diagnostic Workup for Correct Diagnosis of Advanced Systemic Mastocytosis. J. Allergy Clin. Immunol. Pr. 2020, 8, 3121–3127. [Google Scholar] [CrossRef] [PubMed]
- Incorvaia, C.; Frati, F.; Dell’Albani, I.; Robino, A.; Cattaneo, E.; Mauro, M.; David, M.; Qualizza, R.; Pastorello, E. Safety of Hymenoptera Venom Immunotherapy: A Systematic Review. Expert Opin. Pharmacother. 2011, 12, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Ruëff, F.; Chatelain, R.; Przybilla, B. Management of Occupational Hymenoptera Allergy. Curr. Opin. Allergy Clin. Immunol. 2011, 11, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, P.; Gonzalez-de-Olano, D.; Zanotti, R.; Riccio, A.; De Ferrari, L.; Lombardo, C.; Rogkakou, A.; Escribano, L.; Alvarez-Twose, I.; Matito, A.; et al. Venom Immunotherapy in Patients with Clonal Mast Cell Disorders: Efficacy, Safety, and Practical Considerations. J. Allergy Clin. Immunol. Pract. 2013, 1, 474–478. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Systemic Mastocytosis | Lim et al. [10] n (%) | Sanchez et al. [22] n (%) | Wimazal et al. [23] n (%) | Cohen et al. [11] n (%) | Pieri et al. [24] n (%) | Sperr et al. [13] n (%) | GISM Veneto (2021) | GISM Verona (2021) | Veneto vs. Verona (p) |
|---|---|---|---|---|---|---|---|---|---|
| ISMs+ n° | 159 | 93 | 81 | 450 | 418 | 1006 | 392 | 127 | ns |
| % | (46.5) | (82.3) | (82.6) | (82.1) | (89.1) | (76.3) | (91.0) | (89.4) | |
| BMM n° | 36 | 16 | nr | nr | 165 | 268 | 236 | 77 | ns |
| % | (10.5) | (14.1) | (35.9) | (17.4) | (54.8) | (54.2) | |||
| SSM n° | 22 | nr | 7 | nr | 20 | 53 | 6 | 3 | ns |
| % | (6.4) | (7.1) | (4.3) | (4.0) | (1.4) | (2.1) | |||
| ASM n° | 41 | 11 | 5 | 8 | 28 | 62 | 6 | 2 | ns |
| % | (12.0) | (9.7) | (5.1) | (1.4) | (6.1) | (4.7) | (1.4) | (1.4) | |
| SM-AHN n° | 138 | 6 | 11 | 24 | 21 | 174 | 27 | 10 | ns |
| % | (40.4) | (5.3) | (11.2) | (4.4) | (4.6) | (13.2) | (6.3) | (7.0) | |
| MCL n° | 4 | 2 | 1 | 5 | 1 | 23 | 0 | 0 | ns |
| % | (1.2) | (1.8) | (1.0) | (0.9) | (0.2) | (1.7) | (0) | (0) | |
| total | 342 | 113 | 98 | 548 | 460 | 1318 | 431 | 142 |
| Final Diagnosis | CM | MIS | MMAS | BMM | ISMs+ | SSM | ASM | SM-AHN |
|---|---|---|---|---|---|---|---|---|
| Total cases | 19 | 23 | 29 | 236 | 156 | 6 | 6 | 27 |
| Median age, years (range) | 43 (22–74) | 29 (18–69) | 54 (32–74) | 57 (20–86) | 44 (18–80) | 64 (46–79) | 65 (51–76) | 69 (35–80) |
| Male/Female ratio | 0.73 | 0.77 | 4.8 | 2.0 | 0.90 | 1.0 | 2.0 | 1.5 |
| Skin involvement n° (%) | 19 (100) | 23 (100) | 0 (0) | 0 (0) | 156 (100) | 6 (100) | 2 (33.3) | 9 (33.3) |
| Serum Tryptase | ||||||||
| Median (ng/mL) | 7.5 | 4.3 | 11.0 | 17.4 | 25.5 | 266.5 | 185.0 | 28.0 |
| range | 4.4–14.6 | (2.4–68) | (3.4–27) | (3.6–134) | (2.9–582) | (168–377) | (102–589) | (7.9–761) |
| >20 ng/mL (%) | 0 (0) | 1 (4.3) | 3 (10.3) | 93 (39.4) | 91 (58.3) | 6 (100) | 6 (100) | 17 (63.0) |
| Major WHO SM criteria n° (%) | 0 (0) | nd | 0(0) | 85 (36) | 88 (56) | 6 (100) | 6 (100) | 18 (67) |
| Exon 816 KIT Mutation, n° (%) | 0 (0) | nd | 15 (52) | 224 (95) | 148 (95) | 6 (100) | 4/5 (80) | 18/23 (78) |
| CD25+ and/or CD2+ BM MC; n° (%) | 0 (0) | nd | 15 (52) | 215 (91) | 127 (81) | 6 (100) | 4/5 (80) | 18/23 (67) |
| >25% BM atypical MCs; n° (%) | 0 (0) | nd | 9 (31) | 204 (86) | 122 (78) | 3/6 (50) | 5/5 (100) | 17/25 (68) |
| Organomegaly n° (%) | ||||||||
| 1 (0) | 0 (0) | 1 (3) | 5 (2) | 6 (4) | 2 (33) | 5 (83) | 13 (48) |
| 0 (0) | 0 (0) | 0 (0) | 14 (6) | 12 (8) | 0 (0) | 3 (50) | 9 (33) |
| 0 (0) | 2 (9) | 0 (0) | 1(0,4) | 2 (1) | 1 (17) | 2 (33) | 7 (26) |
| Final Diagnosis | CM | MIS | MMAS | BMM | ISMs+ | SSM | ASM | SM-AHN |
|---|---|---|---|---|---|---|---|---|
| HVA, n° (%) | 1 (5) | 2 (9) | 25 (86) | 184 (78) | 34 (22) | 0 (0) | 1 (17) | 4 (15) |
| Other insects n° (%) | 0 (0) | 0 (0) | 1 (3.4) | 3 (1.3) | 2 (1.3) | 1 (17) | 0 (0) | 0 (0) |
| Drug, n° (%) | 1 (5) | 1 (4) | 0 (0) | 22 (9) | 20 (13) | 1 (17) | 2 (33) | 6 (22) |
| Food, n° (%) | 0 (0) | 3 (13) | 1 (3) | 14 (6) | 6 (4) | 0 (0) | 0 (0) | 0 (0) |
| Type of Bone Involvement | Total n° (%) | Median Age, y (Range) | Males >50 y n° (%) | Females >50 y n° (%) | p-Value (Males vs. Females) >50 y | Males ≤ 50 y n° (%) | Females ≤50 y n° (%) | p-Value (Males vs. Females) ≤50 y |
|---|---|---|---|---|---|---|---|---|
| Evaluated patients | 459 | 162 | 103 | 113 | 81 | |||
| Osteopenia | 147 (32) | 52 (20–80) | 50 (31) | 30 (29) | 0.763 | 39 (35) | 28 (35) | 0.993 |
| Osteoporosis | 161 (35) | 58 (20–86) | 60 (37) | 61 (59) | <0.001 | 27 (24) | 13 (16) | 0.182 |
| Fragility fractures | 107 (23) | 61 (39–86) | 45 (28) | 46 (45) | 0.004 | 14 (12) | 2 (2.5) | 0.013 |
| Osteosclerosis | ||||||||
| 15 (3) | 60 (35–79) | 6 (3.7) | 5 (4.9) | 0.647 | 1 (0.8) | 3 (3.7) | 0.170 |
| 26 (6) | 54 (23–74) | 15 (9.3) | 3 (2.9) | 0.453 | 3 (2.7) | 5 (6.2) | 0.224 |
| Final Diagnosis | CM | MIS | MMAS | BMM | ISMs+ | SSM | ASM | SM-AHN |
|---|---|---|---|---|---|---|---|---|
| Evaluable patients | 19 | 13 | 24 | 223 | 149 | 6 | 5 | 20 |
| Osteopenia | 5 (26) | 3 (23) | 6 (25) | 71 (32) | 57 (39) | 0 (0) | 1 (20) | 4 (19) |
| Osteoporosis | 4 (21) | 4 (31) | 4 (17) | 93 (42) | 43 (29) | 0 (0) | 2 (40) | 11 (55) |
| Fragility fractures | 1 (5) | 1 (8) | 3 (12.5) | 73 (33) | 18 (12) | 1 (17) | 2 (40) | 9 (43) |
| Osteosclerosis | 1 (5) | 1 (8) | 0 (0) | 15 (7) | 13 (9) | 6 (100) | 2 (40) | 3 (14) |
| focal | 1 | 1 | 0 | 13 | 9 | 0 | 1 | 1 |
| diffuse | 0 | 0 | 0 | 2 | 4 | 6 | 1 | 2 |
| Small osteolysis | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 2 (10) |
| Large osteolysis | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanotti, R.; Bonifacio, M.; Isolan, C.; Tanasi, I.; Crosera, L.; Olivieri, F.; Orsolini, G.; Schena, D.; Bonadonna, P. A Multidisciplinary Diagnostic Approach Reveals a Higher Prevalence of Indolent Systemic Mastocytosis: 15-Years’ Experience of the GISM Network. Cancers 2021, 13, 6380. https://doi.org/10.3390/cancers13246380
Zanotti R, Bonifacio M, Isolan C, Tanasi I, Crosera L, Olivieri F, Orsolini G, Schena D, Bonadonna P. A Multidisciplinary Diagnostic Approach Reveals a Higher Prevalence of Indolent Systemic Mastocytosis: 15-Years’ Experience of the GISM Network. Cancers. 2021; 13(24):6380. https://doi.org/10.3390/cancers13246380
Chicago/Turabian StyleZanotti, Roberta, Massimiliano Bonifacio, Cecilia Isolan, Ilaria Tanasi, Lara Crosera, Francesco Olivieri, Giovanni Orsolini, Donatella Schena, and Patrizia Bonadonna. 2021. "A Multidisciplinary Diagnostic Approach Reveals a Higher Prevalence of Indolent Systemic Mastocytosis: 15-Years’ Experience of the GISM Network" Cancers 13, no. 24: 6380. https://doi.org/10.3390/cancers13246380
APA StyleZanotti, R., Bonifacio, M., Isolan, C., Tanasi, I., Crosera, L., Olivieri, F., Orsolini, G., Schena, D., & Bonadonna, P. (2021). A Multidisciplinary Diagnostic Approach Reveals a Higher Prevalence of Indolent Systemic Mastocytosis: 15-Years’ Experience of the GISM Network. Cancers, 13(24), 6380. https://doi.org/10.3390/cancers13246380