
TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment




{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. The Tumor Metabolic Reprogramming
1.2. Transforming Growth Factor Beta Signaling in Tumors
2. TGF-β Induced Cancer-Associated Fibroblast (CAF) Metabolic Reprogramming
2.1. CAF General Function
2.2. How Does TGF-β Directly Alter CAF Functions?
2.3. Does the TGF-β Dependent CAF Activity Shape the Tumor Metabolism?
3. TGF-β Induced Endothelial Metabolic Reprogramming
3.1. Endothelial Cell (EC) General Function
3.2. How Does TGF-β Directly Alter EC Metabolic Reprogramming?
3.3. Does the EC Activity Shape the Tumor Metabolism?
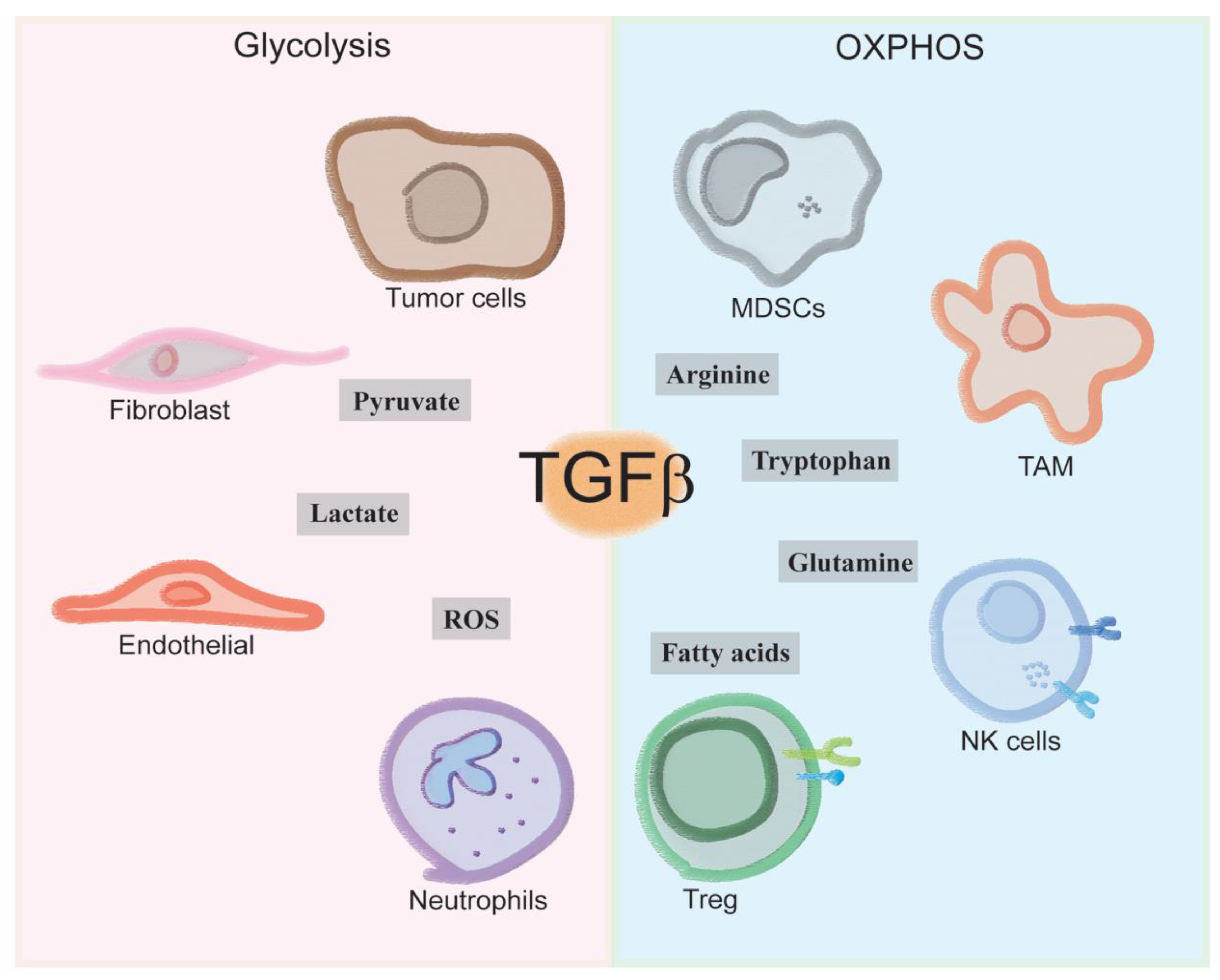
4. Immune Cells Tgfb-Induced Metabolism
4.1. How Does TGF-β Make-Up the Immune Landscape of the Tumor Microenvironment?
4.2. How Does Immune-Metabolism Shape the Tumor Microenvironment?
4.3. How Does TGF-β Influence Immune-Metabolism in the Tumor Microenvironment?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lau, A.N.; Vander Heiden, M.G. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Zhao, F.; Thompson, C.B. The Molecular Determinants of de Novo Nucleotide Biosynthesis in Cancer Cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of Hypoxia-Inducible Factor 1alpha in Common Human Cancers and Their Metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Dang, C.V. C-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell 2018, 33, 368–385.e7. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Massagué, J. Contextual Determinants of TGFβ Action in Development, Immunity and Cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β Structure and Activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.F.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 Is a Major Activator of TGF-Β1 In Vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) Is Essential for the Surface Expression of Latent TGF-Beta on Platelets and Activated FOXP3+ Regulatory T Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Garrison, B.S.; Ma, W.; Wang, R.; Jiang, A.; Li, J.; Mistry, M.; Bronson, R.T.; Santoro, D.; Franco, C.; et al. A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System. Cell 2018, 174, 156–171.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Kim, H.; Liu, X.; Sugiura, H.; Kohyama, T.; Fang, Q.; Wen, F.-Q.; Abe, S.; Wang, X.; Atkinson, J.J.; et al. Matrix Metalloproteinase-9 Activates TGF-β and Stimulates Fibroblast Contraction of Collagen Gels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L1006–L1015. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.J.; Sizeland, A.M. A Pivotal Role for the Transmembrane Domain in Transforming Growth Factor-Beta Receptor Activation. J. Biol. Chem. 1999, 274, 11773–11781. [Google Scholar] [CrossRef] [Green Version]
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan Presents Ligand to the TGFβ Signaling Receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural Determinants of Smad Function in TGF-β Signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Edlund, S.; Landström, M.; Heldin, C.-H.; Aspenström, P. Transforming Growth Factor-Beta-Induced Mobilization of Actin Cytoskeleton Requires Signaling by Small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 Transcriptional Repressor Complex Promotes TGF-Beta Mediated Epithelial-Mesenchymal Transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Derynck, R. Cell Size and Invasion in TGF-β–Induced Epithelial to Mesenchymal Transition Is Regulated by Activation of the MTOR Pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, R.F.; Pöyry, T.A.A.; Stoneley, M.; Willis, A.E. Signaling from MTOR to EIF2α Mediates Cell Migration in Response to the Chemotherapeutic Doxorubicin. Sci. Signal. 2019, 12, eaaw6763. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P. Metabolic Exchanges within Tumor Microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer Associated Fibroblasts: The Dark Side of the Coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of Fibroblast Heterogeneity in the Tumor Microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers 2019, 11, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.S.; Huvos, A.G.; Casas-Ganem, J.E.; Yang, R.; Linkov, I.; Sowers, R.; DiResta, G.R.; Gorlick, R.; Healey, J.H. Tumor Interstitial Fluid Pressure May Regulate Angiogenic Factors in Osteosarcoma. J. Orthop. Res. 2008, 26, 1520–1525. [Google Scholar] [CrossRef]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and Biochemical Remodeling of Stromal Extracellular Matrix in Cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and Its Microenvironment: A Synergistic Interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF Beta1 Induces Prolonged Mitochondrial ROS Generation through Decreased Complex IV Activity with Senescent Arrest in Mv1Lu Cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.-O.; Jung, H.-J.; Seo, Y.-H.; Lee, Y.-K.; Hwang, S.-C.; Hwang, E.S.; Yoon, G. GSK3 Inactivation Is Involved in Mitochondrial Complex IV Defect in Transforming Growth Factor (TGF) Β1-Induced Senescence. Exp. Cell Res. 2012, 318, 1808–1819. [Google Scholar] [CrossRef]
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-β Regulates Nox4, MnSOD and Catalase Expression, and IL-6 Release in Airway Smooth Muscle Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 300, L295–L304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.-M.; Vayalil, P.K.; Ballinger, C.; Dickinson, D.A.; Huang, W.-T.; Wang, S.; Kavanagh, T.J.; Matthews, Q.L.; Postlethwait, E.M. Transforming Growth Factor β Suppresses Glutamate-Cysteine Ligase Gene Expression and Induces Oxidative Stress in A Lung Fibrosis Model. Free Radic. Biol. Med. 2012, 53, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. BioMed Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor Cells Induce the Cancer Associated Fibroblast Phenotype via Caveolin-1 Degradation: Implications for Breast Cancer and DCIS Therapy with Autophagy Inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Zhang, J.; Wang, Y.; Xiong, W.; Mi, J. TGFBR-IDH1-Cav1 Axis Promotes TGF-β Signalling in Cancer-Associated Fibroblast. Oncotarget 2017, 8, 83962–83974. [Google Scholar] [CrossRef] [Green Version]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and Cancer Metabolism in the Tumor Microenvironment: Markers, Models, and Mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467. [Google Scholar] [CrossRef]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 Is A Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.; Lin, Z.; Witkiewicz, A.; et al. Oxidative Stress in Cancer Associated Fibroblasts Drives Tumor-Stroma Co-Evolution. Cell Cycle 2010, 9, 3276–3296. [Google Scholar] [CrossRef] [Green Version]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and Mitochondrial SOD2 (MnSOD) Function as Tumor Suppressors in the Stromal Microenvironment: A New Genetically Tractable Model for Human Cancer Associated Fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of Oxidative Stress and the Microenvironment in Breast Cancer Development and Progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [PubMed] [Green Version]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-Beta in CAF-Mediated Tumor Growth and Metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and Senescence in Cancer-Associated Fibroblasts Metabolically Supports Tumor Growth and Metastasis via Glycolysis and Ketone Production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent Stroma Promotes Prostate Cancer Progression: The Role of MiR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal–Epithelial Metabolic Coupling in Cancer: Integrating Autophagy and Metabolism in the Tumor Microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The Reverse Warburg Effect Is Likely to Be an Achilles’ Heel of Cancer That Can Be Exploited for Cancer Therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate Transporter 4 (MCT4) Is a High Affinity Transporter Capable of Exporting Lactate in High-Lactate Microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernández-Pérez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-Cell-Autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865.e6. [Google Scholar] [CrossRef] [Green Version]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone Bodies and Two-Compartment Tumor Metabolism: Stromal Ketone Production Fuels Mitochondrial Biogenesis in Epithelial Cancer Cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaGory, E.L.; Giaccia, A.J. The Ever Expanding Role of HIF in Tumour and Stromal Biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, S.J.; García-Caballero, M.; Carmeliet, P. Metabolic Signatures of Distinct Endothelial Phenotypes. Trends Endocrinol. Metab. 2020, 31, 580–595. [Google Scholar] [CrossRef]
- Betz, C.; Lenard, A.; Belting, H.-G.; Affolter, M. Cell Behaviors and Dynamics during Angiogenesis. Development 2016, 143, 2249–2260. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.-M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bennewith, K.L.; Durand, R.E. Quantifying Transient Hypoxia in Human Tumor Xenografts by Flow Cytometry. Cancer Res. 2004, 64, 6183–6189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The Hypoxic Tumour Microenvironment. Oncogenesis 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy Metabolism of Cancer: Glycolysis versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecchin, A.; Kalucka, J.; Dubois, C.; Carmeliet, P. How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Front. Immunol. 2017, 8, 1750. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis Revisited from a Metabolic Perspective: Role and Therapeutic Implications of Endothelial Cell Metabolism. Open Biol. 2017, 7, 170219. [Google Scholar] [CrossRef] [Green Version]
- Coomans de Brachène, A.; Demoulin, J.-B. FOXO Transcription Factors in Cancer Development and Therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 Couples Metabolic Activity and Growth State in the Vascular Endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.; Zhao, Y.; Ye, Y.; Yu, J. Opposing Roles and Potential Antagonistic Mechanism between TGF-β and BMP Pathways: Implications for Cancer Progression. EBioMedicine 2019, 41, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The Perivascular Niche Regulates Breast Tumor Dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Prunier, C.; Baker, D.; ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-Mesenchymal Transition in Cancer. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming Growth Factor-Β2 Promotes Snail-Mediated Endothelial–Mesenchymal Transition through Convergence of Smad-Dependent and Smad-Independent Signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail Is Required for TGFβ-Induced Endothelial-Mesenchymal Transition of Embryonic Stem Cell-Derived Endothelial Cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [Green Version]
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-Induced Pulmonary Hypertension through Transforming Growth Factor-β–Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2017, 58, 194–207. [Google Scholar] [CrossRef]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.-X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.-S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Verdegem, D.; Moens, S.; Stapor, P.; Carmeliet, P. Endothelial Cell Metabolism: Parallels and Divergences with Cancer Cell Metabolism. Cancer Metab. 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Cantelmo, A.R.; Conradi, L.-C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.-A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease: Impact of Hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-ΚB/IL-8 Pathway That Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, G.-X.; Kazlauskas, A. Lactate Engages Receptor Tyrosine Kinases Axl, Tie2, and Vascular Endothelial Growth Factor Receptor 2 to Activate Phosphoinositide 3-Kinase/Akt and Promote Angiogenesis. J. Biol. Chem. 2013, 288, 21161–21172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-M.; Kang, M.; Jang, Y.J.; Yang, S.-J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, E.; Angioni, R.; Molon, B.; Calì, B. Chemokines and Chemokine Receptors: Orchestrating Tumor Metastasization. Int. J. Mol. Sci. 2018, 20, 96. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.T.; Luster, A.D. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Dranoff, G. Cytokines in Cancer Pathogenesis and Cancer Therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The Polarization of Immune Cells in the Tumour Environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal Gene Expression Defines Poor-Prognosis Subtypes in Colorectal Cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shen, C.; Wang, X.; Lai, Y.; Zhou, K.; Li, P.; Liu, L.; Che, G. Prognostic Value of TGF-β in Lung Cancer: Systematic Review and Meta-Analysis. BMC Cancer 2019, 19, 691. [Google Scholar] [CrossRef] [Green Version]
- De Kruijf, E.M.; Dekker, T.J.A.; Hawinkels, L.J.A.C.; Putter, H.; Smit, V.T.H.B.M.; Kroep, J.R.; Kuppen, P.J.K.; van de Velde, C.J.H.; ten Dijke, P.; Tollenaar, R.A.E.M.; et al. The Prognostic Role of TGF-β Signaling Pathway in Breast Cancer Patients. Ann. Oncol. 2013, 24, 384–390. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Immune-Mediated Eradication of Tumors through the Blockade of Transforming Growth Factor-Beta Signaling in T Cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-β Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T Cells Suppress Tumor-Specific CD8 T Cell Cytotoxicity through TGF-Beta Signals in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting Edge: TGF-Beta Induces A Regulatory Phenotype in CD4+CD25- T Cells through Foxp3 Induction and down-Regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [Green Version]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β Targeted Cancer Therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moo-Young, T.A.; Larson, J.W.; Belt, B.A.; Tan, M.C.; Hawkins, W.G.; Eberlein, T.J.; Goedegebuure, P.S.; Linehan, D.C. Tumor-Derived TGF-Beta Mediates Conversion of CD4+Foxp3+ Regulatory T Cells in A Murine Model of Pancreas Cancer. J. Immunother. 2009, 32, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Downs-Canner, S.; Berkey, S.; Delgoffe, G.M.; Edwards, R.P.; Curiel, T.; Odunsi, K.; Bartlett, D.L.; Obermajer, N. Suppressive IL-17A+Foxp3+ and Ex-Th17 IL-17AnegFoxp3+ Treg Cells Are A Source of Tumour-Associated Treg Cells. Nat. Commun. 2017, 8, 14649. [Google Scholar] [CrossRef]
- Oh, E.; Hong, J.; Yun, C.-O. Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial–Mesenchymal Transition. Cells 2019, 8, 1387. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.Y.; Flavell, R.A. “Yin-Yang” Functions of Transforming Growth Factor-Beta and T Regulatory Cells in Immune Regulation. Immunol. Rev. 2007, 220, 199–213. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Tsunawaki, S.; Sporn, M.; Ding, A.; Nathan, C. Deactivation of Macrophages by Transforming Growth Factor-β. Nature 1988, 334, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of TGF-β Paradox in Benign and Malignant Cells. Front. Oncol. 2014, 4, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-Associated Macrophages Are A Distinct M2 Polarised Population Promoting Tumour Progression: Potential Targets of Anti-Cancer Therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. TGF-Beta and Cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Kuick, R.; Bhan, U.; Chen, J.; Newstead, M.; Keshamouni, V.G. TGF-β-Induced IRAK-M Expression in Tumor-Associated Macrophages Regulates Lung Tumor Growth. Oncogene 2011, 30, 2475–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yang, Y.; Cui, Y.; Wang, C.; Lai, Z.; Li, Y.; Zhang, W.; Mustonen, H.; Puolakkainen, P.; Ye, Y.; et al. Tumor-Associated Macrophages Regulate Gastric Cancer Cell Invasion and Metastasis through TGFβ2/NF-ΚB/Kindlin-2 Axis. Chin. J. Cancer Res. 2020, 32, 72–88. [Google Scholar] [CrossRef]
- Shen, J.; Li, J.; Wang, B.; Jin, H.; Wang, M.; Zhang, Y.; Yang, Y.; Im, H.-J.; O’Keefe, R.; Chen, D. Deletion of the Transforming Growth Factor β Receptor Type II Gene in Articular Chondrocytes Leads to a Progressive Osteoarthritis-like Phenotype in Mice. Arthritis Rheum. 2013, 65, 3107–3119. [Google Scholar] [CrossRef] [Green Version]
- Bonde, A.-K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral Macrophages Contribute to Epithelial-Mesenchymal Transition in Solid Tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil Diversity and Plasticity in Tumour Progression and Therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Allen, S.S.; Mackie, J.T.; Russell, K.; Jeevan, A.; Skwor, T.A.; McMurray, D.N. Altered Inflammatory Responses Following Transforming Growth Factor-Beta Neutralization in Experimental Guinea Pig Tuberculous Pleurisy. Tuberclosis 2008, 88, 430–436. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Liu, X.; Chen, J.; Huang, S.; Wei, W.; Zou, Y.; Liu, X.; Deng, K.; Mo, S.; Chen, J.; et al. Anti-TGF-β Attenuates Tumor Growth via Polarization of Tumor Associated Neutrophils towards an Anti-Tumor Phenotype in Colorectal Cancer. J. Cancer 2020, 11, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Germann, M.; Zangger, N.; Sauvain, M.-O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils Suppress Tumor-Infiltrating T Cells in Colon Cancer via Matrix Metalloproteinase-Mediated Activation of TGFβ. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Solito, S.; Ugel, S.; Molon, B.; Bronte, V.; Marigo, I. MDSCs in Cancer: Conceiving New Prognostic and Therapeutic Targets. Biochim. Biophys. Acta 2016, 1865, 35–48. [Google Scholar] [CrossRef]
- Trikha, P.; Carson, W.E. Signaling Pathways Involved in MDSC Regulation. Biochim. Biophys. Acta 2014, 1846, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 Is Required for the Accumulation and Functions of Tumor-Expanded Myeloid-Derived Suppressor Cells via Targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef]
- Lee, C.-R.; Lee, W.; Cho, S.K.; Park, S.-G. Characterization of Multiple Cytokine Combinations and TGF-β on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-Β1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.G.; Liebertz, D.J.; Epstein, A.L. Characterization of Cytokine-Induced Myeloid-Derived Suppressor Cells from Normal Human Peripheral Blood Mononuclear Cells. J. Immunol. 2010, 185, 2273–2284. [Google Scholar] [CrossRef]
- Jayaraman, P.; Parikh, F.; Newton, J.M.; Hanoteau, A.; Rivas, C.; Krupar, R.; Rajapakshe, K.; Pathak, R.; Kanthaswamy, K.; MacLaren, C.; et al. TGF-Β1 Programmed Myeloid-Derived Suppressor Cells (MDSC) Acquire Immune-Stimulating and Tumor Killing Activity Capable of Rejecting Established Tumors in Combination with Radiotherapy. OncoImmunology 2018, 7, e1490853. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Orchestration of Metabolism by Macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghesquière, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of Stromal and Immune Cells in Health and Disease. Nature 2014, 511, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, P.J.; Rathmell, J.C. T Cell Metabolic Fitness in Antitumor Immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Molon, B.; Calì, B.; Viola, A. T Cells and Cancer: How Metabolism Shapes Immunity. Front. Immunol. 2016, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is A Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Dimeloe, S.; Gubser, P.; Loeliger, J.; Frick, C.; Develioglu, L.; Fischer, M.; Marquardsen, F.; Bantug, G.R.; Thommen, D.; Lecoultre, Y.; et al. Tumor-Derived TGF-β Inhibits Mitochondrial Respiration to Suppress IFN-γ Production by Human CD4+ T Cells. Sci. Signal. 2019, 12, eaav3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyadharshini, B.; Loschi, M.; Newton, R.H.; Zhang, J.-W.; Finn, K.K.; Gerriets, V.A.; Huynh, A.; Rathmell, J.C.; Blazar, B.R.; Turka, L.A. Cutting Edge: TGF-β and Phosphatidylinositol 3-Kinase Signals Modulate Distinct Metabolism of Regulatory T Cell Subsets. J. Immunol. 2018, 201, 2215–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiatz-Bittencourt, V.; Finlay, D.K.; Gardiner, C.M. Canonical TGF-β Signaling Pathway Represses Human NK Cell Metabolism. J. Immunol. 2018, 200, 3934–3941. [Google Scholar] [CrossRef]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the MTOR Pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming Growth Factor Beta 1 Inhibits Expression of NKp30 and NKG2D Receptors: Consequences for the NK-Mediated Killing of Dendritic Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-Beta1 Secretion and down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Van Belle, T.L.; von Herrath, M.G. The Role of the Activating Receptor NKG2D in Autoimmunity. Mol. Immunol. 2009, 47, 8–11. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Takami, A.; Yoshioka, K.; Nakata, K.; Sato, T.; Kasahara, Y.; Nakao, S. Human MicroRNA-1245 down-Regulates the NKG2D Receptor in Natural Killer Cells and Impairs NKG2D-Mediated Functions. Haematologica 2012, 97, 1295–1303. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-Activated Macrophages Induced Aerobic Glycolysis and Epithelial-Mesenchymal Transition in Breast Cancer by Regulation of CCL5-CCR5 Axis: A Positive Metabolic Feedback Loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef] [Green Version]
- Gómez, V.; Eykyn, T.R.; Mustapha, R.; Flores-Borja, F.; Male, V.; Barber, P.R.; Patsialou, A.; Green, R.; Panagaki, F.; Li, C.W.; et al. Breast Cancer–Associated Macrophages Promote Tumorigenesis by Suppressing Succinate Dehydrogenase in Tumor Cells. Sci. Signal. 2020, 13, eaax4585. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punzo, F.; Bellini, G.; Tortora, C.; Pinto, D.D.; Argenziano, M.; Pota, E.; Paola, A.D.; Martino, M.D.; Rossi, F. Mifamurtide and TAM-like Macrophages: Effect on Proliferation, Migration and Differentiation of Osteosarcoma Cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; Gao, X.; et al. Hypoxia Promotes Glioma-Associated Macrophage Infiltration via Periostin and Subsequent M2 Polarization by Upregulating TGF-Beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Rodriguez, P.C.; Ochoa, A.C. Energy Metabolic Pathways Control the Fate and Function of Myeloid Immune Cells. J. Leukoc. Biol. 2017, 102, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Klein Geltink, R.I.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine Nitration Prevents Intratumoral Infiltration of Antigen-Specific T Cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-Derived Suppressor Cells Coming of Age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression Mediated by Myeloid-Derived Suppressor Cells (MDSCs) during Tumour Progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angioni, R.; Liboni, C.; Herkenne, S.; Sánchez-Rodríguez, R.; Borile, G.; Marcuzzi, E.; Calì, B.; Muraca, M.; Viola, A. CD73+ Extracellular Vesicles Inhibit Angiogenesis through Adenosine A2B Receptor Signalling. J. Extracell. Vesicles 2020, 9, 1757900. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 Upregulation on Myeloid-Derived Suppressor Cells via TGF-β-MTOR-HIF-1 Signaling in Patients with Non-Small Cell Lung Cancer. OncoImmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [Green Version]
- De Sanctis, F.; Sandri, S.; Ferrarini, G.; Pagliarello, I.; Sartoris, S.; Ugel, S.; Marigo, I.; Molon, B.; Bronte, V. The Emerging Immunological Role of Post-Translational Modifications by Reactive Nitrogen Species in Cancer Microenvironment. Front. Immunol. 2014, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Dzik, J.M. Evolutionary Roots of Arginase Expression and Regulation. Front. Immunol. 2014, 5, 544. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered Recognition of Antigen Is a Mechanism of CD8 + T Cell Tolerance in Cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular Mechanisms of Neutrophil Dysfunction in Glycogen Storage Disease Type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; de Melo, M.P. Glutamine Metabolism by Lymphocytes, Macrophages, and Neutrophils: Its Importance in Health and Disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef]
- Stanton, R.C. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondo, R.; Barisione, G.; Mastracci, L.; Grossi, F.; Orengo, A.M.; Costa, R.; Truini, M.; Fabbi, M.; Ferrini, S.; Barbieri, O. IL-8 Induces Exocytosis of Arginase 1 by Neutrophil Polymorphonuclears in Nonsmall Cell Lung Cancer. Int. J. Cancer 2009, 125, 887–893. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angioni, R.; Sánchez-Rodríguez, R.; Viola, A.; Molon, B. TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment. Cancers 2021, 13, 401. https://doi.org/10.3390/cancers13030401
Angioni R, Sánchez-Rodríguez R, Viola A, Molon B. TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment. Cancers. 2021; 13(3):401. https://doi.org/10.3390/cancers13030401
Chicago/Turabian StyleAngioni, Roberta, Ricardo Sánchez-Rodríguez, Antonella Viola, and Barbara Molon. 2021. "TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment" Cancers 13, no. 3: 401. https://doi.org/10.3390/cancers13030401