
DNA Damage Response in Multiple Myeloma: The Role of the Tumor Microenvironment



Abstract
Simple Summary
Abstract
1. Introduction
2. Genomic Damage
3. Genomic Instability in MM
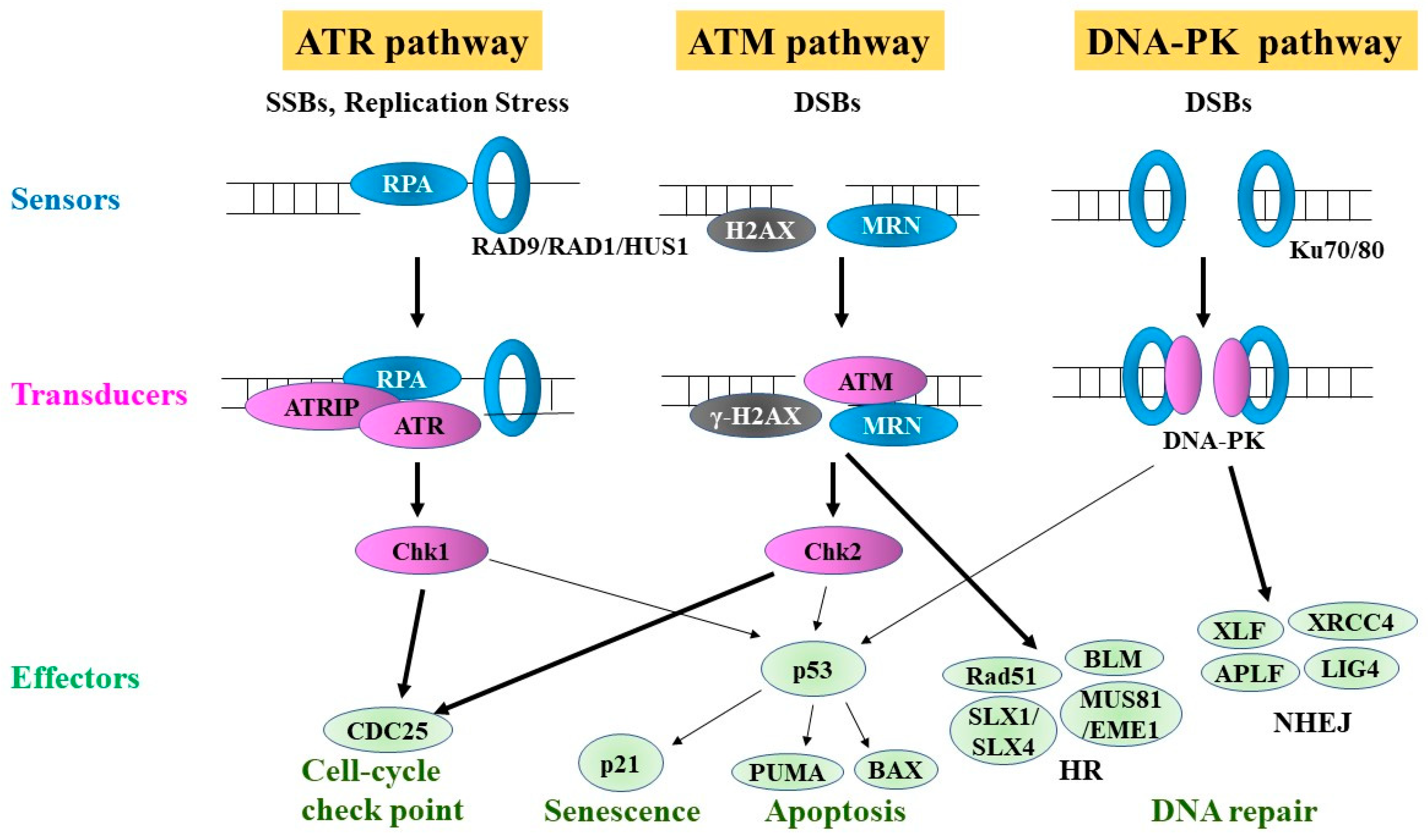
4. DDR
5. DNA Repair Pathways
6. Major Single-Strand Break (SSB) Repair Pathways
6.1. BER Pathway
6.2. NER Pathway
6.3. MMR Pathway
7. DSB Repair Pathways
7.1. HRR Pathway
7.2. NHEJ Pathway
7.3. DSB Repair Pathways and MM
8. FA Pathway
9. Epigenetic Machinery and DNA Damage
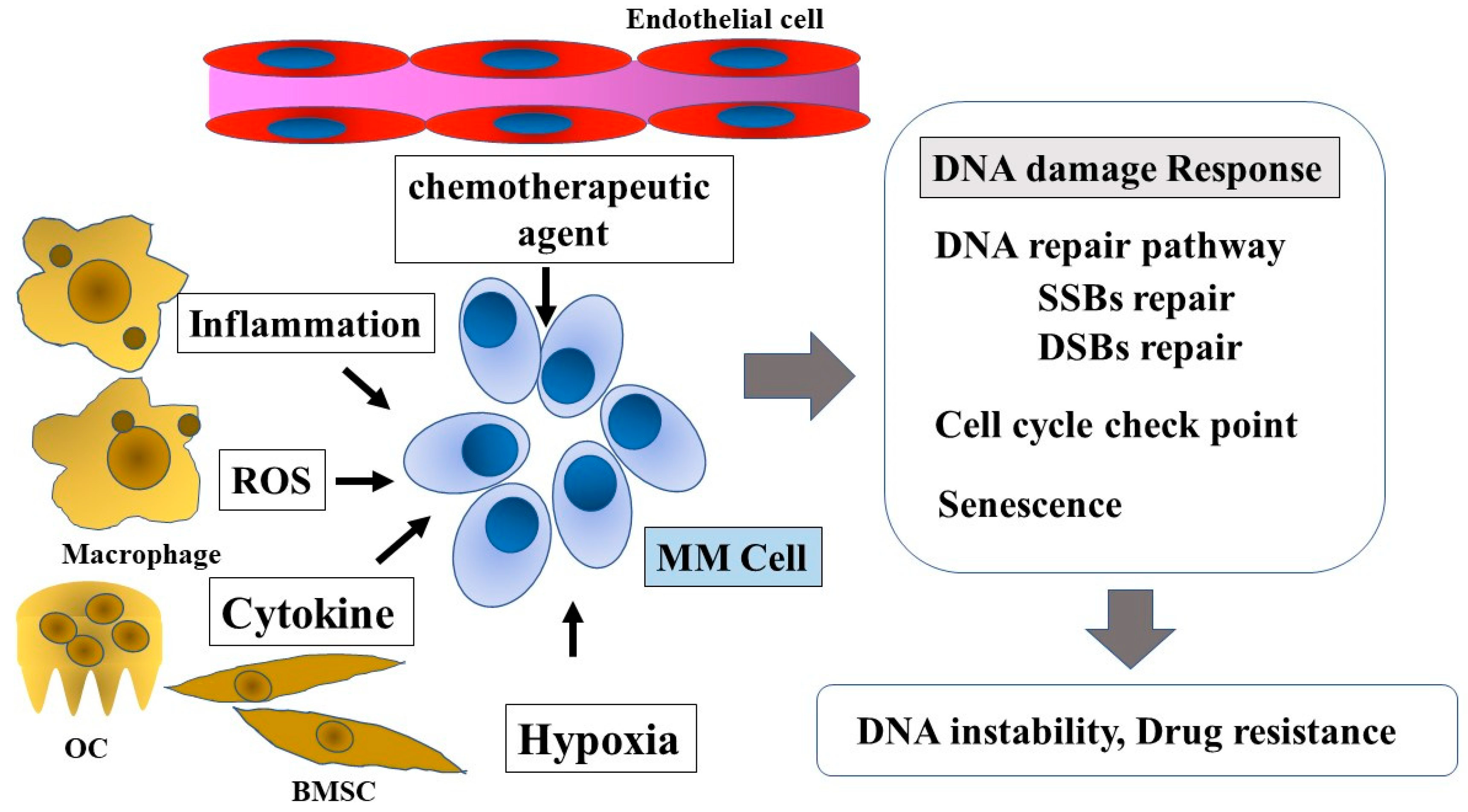
10. Inflammatory Microenvironment and ROS
11. Hypoxia
12. Cellular Metabolites
13. Different DDR Pathways and Their Associated Inhibitors in MM
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Bergsagel, D.E.; Sprague, C.C.; Austin, C.; Griffith, K.M. Evaluation of new chemotherapeutic agents in the treatment of multiple myeloma. IV. L-Phenylalanine mustard (NSC-8806). Cancer Chemother. Rep. 1962, 21, 87–99. [Google Scholar]
- Osgood, E.E. The survival time of patients with plasmocytic myeloma. Cancer Chemother. Rep. 1960, 9, 1–10. [Google Scholar]
- Attal, M.; Harousseau, J.L.; Stoppa, A.M.; Sotto, J.J.; Fuzibet, J.G.; Rossi, J.F.; Casassus, P.; Maisonneuve, H.; Facon, T.; Ifrah, N.; et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Français du Myélome. N. Engl. J. Med. 1996, 335, 91–97. [Google Scholar] [CrossRef]
- Child, J.A.; Morgan, G.J.; Davies, F.E.; Owen, R.G.; Bell, S.E.; Hawkins, K.; Brown, J.; Drayson, M.T.; Selby, P.J. Medical Research Council Adult Leukaemia Working Party. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N. Engl. J. Med. 2003, 348, 1875–1883. [Google Scholar] [CrossRef]
- Durie, B.G.M.; Hoering, A.; Abidi, M.H.; Rajkumar, S.V.; Epstein, J.; Kahanic, S.P.; Thakuri, M.; Reu, F.; Reynolds, C.M.; Sexton, R.; et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): A randomised, open-label, phase 3 trial. Lancet 2017, 389, 519–527. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J. Clin. Oncol. 2009, 27, 5008–5014. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. TOURMALINE-MM1 Study Group. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. ELOQUENT-2 Investigators. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. ICARIA-MM study group. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinex-or-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Shammas, M.A.; Shmookler Reis, R.J.; Koley, H.; Batchu, R.B.; Li, C.; Munshi, N.C. Dysfunctional homologous recombination medi-ates genomic instability and progression in myeloma. Blood 2009, 113, 2290–2297. [Google Scholar] [CrossRef]
- Walters, D.K.; Wu, X.; Tschumper, R.C.; Arendt, B.K.; Huddleston, P.M.; Henderson, K.J.; Dispenzieri, A.; Jelinek, D.F. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia 2011, 25, 1344–1353. [Google Scholar] [CrossRef]
- Herrero, A.B.; San Miguel, J.; Gutierrez, N.C. Deregulation of DNA double-strand break repair in multiple myeloma: Implications for genome stability. PLoS ONE 2015, 10, e0121581. [Google Scholar] [CrossRef]
- Cottini, F.; Hideshima, T.; Suzuki, R.; Tai, Y.T.; Bianchini, G.; Richardson, P.G.; Anderson, K.C.; Tonon, G. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015, 5, 972–987. [Google Scholar] [CrossRef]
- Yuan, J.; Glazer, P.M. Mutagenesis induced by the tumor microenvironment. Mutat. Res. 1998, 400, 439–446. [Google Scholar] [PubMed]
- Cagnetta, A.; Lovera, D.; Grasso, R.; Colombo, N.; Canepa, L.; Ballerini, F.; Calvio, M.; Miglino, M.; Gobbi, M.; Lemoli, R.; et al. Mechanisms and Clinical Applications of Genome Instability in Multiple Myeloma. BioMed Res. Int. 2015, 2015, 943096. [Google Scholar] [CrossRef] [PubMed]
- Tshering, D. Lama-Sherpa 1, Lalita A Shevde An Emerging Regulatory Role for the Tumor Microenvironment in the DNA Damage Response to Double-Strand Breaks. Mol. Cancer Res. 2020, 18, 185–193. [Google Scholar]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [PubMed]
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenesis 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagenesis 2014, 5, 1000165. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Chng, W.J.; Fonseca, R. Centrosomes and myeloma; aneuploidy and proliferation. Environ. Mol. Mutagenesis 2009, 50, 697–707. [Google Scholar] [CrossRef]
- Neuse, C.J.; Lomas, O.C.; Schliemann, C.; Shen, Y.J.; Manier, S.; Bustoros, M.; Ghobrial, I.M. Genome instability in multiple myeloma. Leukemia 2020, 34, 2887–2897. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Pihan, G.A.; Wallace, J.; Zhou, Y.; Doxsey, S.J. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003, 63, 1398–1404. [Google Scholar] [PubMed]
- Chng, W.J.; Ahmann, G.J.; Henderson, K.; Santana-Davila, R.; Greipp, P.R.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Kumar, S.; Rajkumar, S.V.; et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood 2006, 107, 3669–3675. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J., Jr. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef]
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res. 2005, 569, 75–85. [Google Scholar] [CrossRef]
- Kumar, S.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Gabrea, A.; Leif Bergsagel, P.; Michael Kuehl, W. Distinguishing primary and secondary translocations in multiple myeloma. DNA Repair 2006, 5, 1225–1233. [Google Scholar] [CrossRef]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.H.; Mulligan, G.; Fonseca, R.; Chng, W.J. A novel measure of chromosome instability can account for prognostic difference in multiple myeloma. PLoS ONE 2013, 8, e66361. [Google Scholar] [CrossRef] [PubMed]
- Spanswick, V.J.; Craddock, C.; Sekhar, M.; Mahendra, P.; Shankaranarayana, P.; Hughes, R.G.; Hochhauser, D.; Hartley, J.A. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood 2002, 100, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Landi, S.; Reiman, T.; Perry, T.; Plesa, A.; Bellini, I.; Barale, R.; Pilarski, L.M.; Troncy, J.; Tavtigian, S.; et al. Genetic polymorphisms associated with outcome in multiple myeloma patients receiving high-dose melphalan. Bone Marrow Transplant. 2010, 45, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Gourzones-Dmitriev, C.; Kassambara, A.; Sahota, S.; Rème, T.; Moreaux, J.; Bourquard, P.; Hose, D.; Pasero, P.; Constantinou, A.; Klein, B. DNA repair pathways in human multiple myeloma: Role in oncogenesis and potential targets for treatment. Cell Cycle 2013, 12, 2760–2773. [Google Scholar] [CrossRef]
- Jacquemont, C.; Taniguchi, T. Proteasome function is required for DNA damage response and fanconi anemia pathway acti-vation. Cancer Res. 2007, 67, 7395–7405. [Google Scholar] [CrossRef]
- Murakawa, Y.; Sonoda, E.; Barber, L.J.; Zeng, W.; Yokomori, K.; Kimura, H.; Niimi, A.; Lehmann, A.; Zhao, G.Y.; Hochegger, H.; et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res. 2007, 67, 8536–8543. [Google Scholar]
- Yarde, D.N.; Oliveira, V.; Mathews, L.; Wang, X.; Villagra, A.; Boulware, D.; Shain, K.H.; Hazlehurst, L.A.; Alsina, M.; Chen, D.T.; et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res. 2009, 69, 9367–9375. [Google Scholar] [CrossRef]
- Popat, R.; Maharaj, L.; Oakervee, H.; Cavenagh, J.; Joel, S. Schedule dependent cytotoxicity of bortezomib and melphalan in multiple myeloma. Br. J. Haematol. 2013, 160, 111–114. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. VISTA Trial Investigators. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008, 359, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Popat, R.; Oakervee, H.; Williams, C.; Cook, M.; Craddock, C.; Basu, S.; Singer, C.; Harding, S.; Foot, N.; Hallam, S.; et al. Bortezomib, low-dose intravenous melphalan, and dexamethasone for patients with relapsed multiple myeloma. Br. J. Haematol. 2009, 144, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.; Moreau, P.; Huynh, A.; Mary, J.-Y.; Danho, C.; Caillot, D.; Hulin, C.; Fruchart, C.; Marit, G.; Pégourié, B.; et al. Intergroupe Francophone du Myélome (IFM) Bortezomib and high-dose melphalan as conditioning regimen before autologous stem cell transplantation in patients with de novo multiple myeloma: A phase 2 study of the Intergroupe Francophone du Myelome (IFM). Blood 2010, 115, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.; Hebraud, B.; Lauwers-Cances, V.; Macro, M.; Leleu, X.; Hulin, C.; Karlin, L.; Royer, B.; Perrot, A.; Moreau, P.; et al. Bortezomib and High-Dose Melphalan vs. High-Dose Melphalan As Conditioning Regimen before Autologous Stem Cell Transplantation in De Novo Multiple Myeloma Patients: A Phase 3 Study of the Intergroupe Francophone Du Myelome (IFM 2014-02). Blood 2017, 130 (Suppl. 1), 398. [Google Scholar]
- Wakharde, A.A.; Awad, A.H.; Bhagat, A.; Karuppayil, S.M. Synergistic Activation of Doxorubicin against Cancer: A Review. Am. J. Clin. Microbiol. Antimicrob. 2018, 1, 1–6. [Google Scholar]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kırdar, B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci. Rep. 2018, 8, 13672. [Google Scholar] [CrossRef]
- Kayser, S.; Döhner, K.; Krauter, J.; Köhne, C.H.; Horst, H.A.; Held, G.; von Lilienfeld-Toal, M.; Wilhelm, S.; Kündgen, A.; Götze, K.; et al. German-Austrian AMLSG. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Singh, N.; Pay, S.L.; Bhandare, S.B.; Arimpur, U.; Motea, E.A. Therapeutic Strategies and Biomarkers to Modulate PARP Activity for Targeted Cancer Therapy. Cancers 2020, 12, 972. [Google Scholar] [CrossRef]
- Gkotzamanidou, M.; Terpos, E.; Bamia, C.; Munshi, N.C.; Dimopoulos, M.A.; Souliotis, V.L. DNA repair of myeloma plasma cells correlates with clinical outcome: The effect of the nonhomologous end-joining inhibitor SCR7. Blood 2016, 128, 1214–1225. [Google Scholar] [CrossRef]
- Herrero, A.B.; Gutiérrez, N.C. Targeting ongoing DNA damage in multiple myeloma: Effects of DNA damage response inhibitors on plasma cell survival. Front. Oncol. 2017, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S. Functional link between DNA damage responses and transcriptional regulation by ATM in response to a histone deacetylase inhibitor TSA. Cancer Res. Treat. 2007, 39, 116–124. [Google Scholar] [CrossRef][Green Version]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxid. Redox Signal. 2013, 19, 559–571. [Google Scholar] [CrossRef]
- Bi, X. Mechanism of DNA damage tolerance. World J. Biol. Chem. 2015, 6, 48–56. [Google Scholar] [CrossRef]
- Yoshiyama, K.O.; Sakaguchi, K.; Kimura, S. DNA damage response in plants: Conserved and variable response compared to animals. Biology 2013, 2, 1338–1356. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Huen, M.S.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res. 2008, 18, 8–16. [Google Scholar]
- Botrugno, O.A.; Bianchessi, S.; Zambroni, D.; Frenquelli, M.; Belloni, D.; Bongiovanni, L.; Girlanda, S.; di Terlizzi, S.; Ferrarini, M.; Ferrero, E.; et al. ATR addiction in multiple myeloma: Synthetic lethal approaches exploiting established therapies. Haematologica 2019, 105, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Landau, H.J.; McNeely, S.C.; Nair, J.S.; Comenzo, R.L.; Asai, T.; Friedman, H.; Jhanwar, S.C.; Nimer, S.D.; Schwartz, G.K. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol. Cancer Ther. 2012, 11, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.P.; Andersen, A.H.; Bjergbæk, L. Abortive activity of Topoisomerase I: A challenge for genome integrity? Curr. Genet. 2019, 65, 1141–1144. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Li, J.; Liu, J.; Baks-Te Bulte, L.; Wiersma, M.; Malik, N.U.; van Marion, D.M.S.; Tolouee, M.; Hoogstra-Berends, F.; et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD+ depletion in experimental atrial fibrillation. Nat. Commun. 2019, 10, 1307. [Google Scholar] [CrossRef]
- Saitoh, T.; Shinmura, K.; Yamaguchi, S.; Tani, M.; Seki, S.; Murakami, H.; Nojima, Y.; Yokota, J. Enhancement of OGG1 protein AP lyase activity by increase of APEX protein. Mutat. Res. 2001, 486, 31–40. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Lin, Y.; Raj, J.; Li, J.; Ha, A.; Hossain, M.A.; Richardson, C.; Mukherjee, P.; Yan, S. APE1 senses DNA single-strand breaks for repair and signaling. Nucleic Acids Res. 2020, 48, 1925–1940. [Google Scholar]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef]
- Thakur, S.; Sarkar, B.; Cholia, R.P.; Gautam, N.; Dhiman, M.; Mantha, A.K. APE1/Ref-1 as an emerging therapeutic target for various human diseases: Phytochemical modulation of its functions. Exp. Mol. Med. 2014, 46, e106. [Google Scholar]
- Schindl, M.; Oberhuber, G.; Pichlbauer, E.G.; Obermair, A.; Birner, P.; Kelley, M.R. DNA repair-redox enzyme apurinic endonuclease in cervical cancer: Evaluation of redox control of HIF-1alpha and prognostic significance. Int. J. Oncol. 2001, 19, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Moore, D.H.; Michael, H.; Kelley, M.R. Studies of apurinic/apyrimidinic endonuclease/ref-1 expression in epithelial ovarian cancer: Correlations with tumor progression and platinum resistance. Clin. Cancer Res. 2013, 9, 4689–4694. [Google Scholar]
- Kelley, M.R.; Cheng, L.; Foster, R.; Tritt, R.; Jiang, J.; Broshears, J.; Koch, M. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin. Cancer Res. 2001, 7, 824–830. [Google Scholar] [PubMed]
- Xie, J.; Zhang, L.; Li, M.; Du, J.; Zhou, L.; Yang, S.; Zeng, L.; Li, Z.; Wang, G.; Wang, D. Functional analysis of the involvement of apurinic/apyrimidinic endonuclease 1 in the resistance to melphalan in multiple myeloma. BMC Cancer 2014, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Talluri, S.; Pal, J.; Yuan, X.; Lu, R.; Nanjappa, P.; Samur, M.K.; Munshi, N.C.; Shammas, M.A. Role of apurinic/apyrimidinic nucleases in the regulation of homologous recombination in myeloma: Mechanisms and translational significance. Blood Cancer J. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. NPJ Precis. Oncol. 2017, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.L.; He, F.; Ye, J.Z.; Wu, H.N.; Zhang, J.Y.; Liu, Z.H.; Li, Y.Q.; Luo, X.L.; Lin, Y.; Liang, R. APE1 overexpression is associated with poor survival in patients with solid tumors: A meta-analysis. Oncotarget 2017, 8, 59720–59728. [Google Scholar] [CrossRef]
- Seo, Y.; Kinsella, T.J. Essential role of DNA base excision repair on survival in an acidic tumor microenvironment. Cancer Res. 2009, 69, 7285–7293. [Google Scholar] [CrossRef]
- Ushie, C.; Saitoh, T.; Iwasaki, A.; Moriyama, N.; Murakami, H. The polymorphisms of base excision repair genes influence the prognosis of multiple myeloma. Blood 2012, 120, 3981. [Google Scholar] [CrossRef]
- Jensen, K.A.; Shi, X.; Yan, S. Genomic alterations and abnormal expression of APE2 in multiple cancers. Sci. Rep. 2020, 10, 3758. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, N.; Minato, Y.; Saitoh, T.; Takahashi, N.; Kasamatsu, T.; Souma, K.; Oda, T.; Hoshino, T.; Sakura, T.; Ishizaki, T.; et al. PARP1 V762A polymorphism affects the prognosis of myelodysplastic syndromes. Eur. J. Haematol. 2020, 104, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; LeDuc, R.D.; Fornelli, L.; Schunter, A.J.; Bennett, R.L.; Kelleher, N.L.; Licht, J.D. Defining the NSD2 interactome: PARP1 PARylation reduces NSD2 histone methyltransferase activity and impedes chromatin binding. J. Biol. Chem. 2019, 294, 12459–12471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Zhao, Y.; Gao, D.; Xing, J.; Liu, H. High PARP-1 expression is associated with tumor invasion and poor prognosis in gastric cancer. Oncol. Lett. 2016, 12, 3825–3835. [Google Scholar]
- Molnár, S.; Beke, L.; Méhes, G.; Póka, R. The Prognostic Value of PARP Expression in High-Grade Epithelial Ovarian Cancer. Pathol. Oncol. Res. 2020, 26, 2549–2555. [Google Scholar] [CrossRef]
- Siraj, A.K.; Pratheeshkumar, P.; Parvathareddy, S.K.; Divya, S.P.; Al-Dayel, F.; Tulbah, A.; Ajarim, D.; Al-Kuraya, K.S. Overexpression of PARP is an independent prognostic marker for poor survival in Middle Eastern breast cancer and its inhibition can be enhanced with embelin co-treatment. Oncotarget 2018, 9, 37319–37332. [Google Scholar] [CrossRef]
- Qin, Q.; Lu, J.; Zhu, H.; Xu, L.; Cheng, H.; Zhan, L.; Yang, X.; Zhang, C.; Sun, X. PARP-1 Val762Ala polymorphism and risk of cancer: A meta-analysis based on 39 case-control studies. PLoS ONE 2014, 9, e98022. [Google Scholar] [CrossRef]
- Yu, H.; Ma, H.; Yin, M.; Wei, Q. Association between PARP-1 V762A polymorphism and cancer susceptibility: A meta-analysis. Genet. Epidemiol. 2012, 36, 56–65. [Google Scholar]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A.; Moore, K.N.; Colombo, N.; Ray-Coquard, I.; Pignata, S. The forefront of ovarian cancer therapy: Update on PARP inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced "BRCAness" sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2020, 106, 185–195. [Google Scholar]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Szalat, R.; Samur, M.K.; Fulciniti, M.; Lopez, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 2018, 32, 111–119. [Google Scholar] [CrossRef]
- Vangsted, A.; Gimsing, P.; Klausen, T.W.; Nexø, B.A.; Wallin, H.; Andersen, P.; Hokland, P.; Lillevang, S.T.; Vogel, U. Polymorphisms in the genes ERCC2, XRCC3 and CD3EAP influence treatment outcome in multiple myeloma patients undergoing autologous bone marrow transplantation. Int. J. Cancer 2007, 120, 1036–1045. [Google Scholar]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [PubMed]
- Gueranger, Q.; Li, F.; Peacock, M.; Larnicol-Fery, A.; Brem, R.; Macpherson, P.; Egly, J.M.; Karran, P. Protein oxidation and DNA repair inhibition by 6-thioguanine and UVA radiation. J. Investig. Dermatol. 2014, 134, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Timurağaoğlu, A.; Demircin, S.; Dizlek, S.; Alanoğlu, G.; Kiriş, E. Microsatellite instability is a common finding in multiple mye-loma. Clin. Lymphoma Myeloma 2009, 9, 371–374. [Google Scholar] [PubMed]
- Velangi, M.R.; Matheson, E.C.; Morgan, G.J.; Jackson, G.H.; Taylor, P.R.; Hall, A.G.; Irving, J.A. DNA mismatch repair pathway defects in the pathogenesis and evolution of myeloma. Carcinogenesis 2004, 25, 1795–1803. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Srivastava, M.; Raghavan, S.C. DNA double-strand break repair inhibitors as cancer therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and com-bination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to dou-ble-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rosidi, B.; Perrault, R.; Wang, M.; Zhang, L.; Windhofer, F.; Iliakis, G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005, 65, 4020–4030. [Google Scholar] [CrossRef]
- Caracciolo, D.; Di Martino, M.T.; Amodio, N.; Morelli, E.; Montesano, M.; Botta, C.; Scionti, F.; Talarico, D.; Altomare, E.; Gallo Cantafio, M.E.; et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia 2019, 33, 487–498. [Google Scholar] [CrossRef]
- Krajewska, M.; Fehrmann, R.S.; de Vries, E.G.; van Vugt, M.A. Regulators of homologous recombination repair as novel targets for cancer treatment. Front. Genet. 2015, 6, 96. [Google Scholar] [CrossRef]
- Tan, D.S.; Rothermundt, C.; Thomas, K.; Bancroft, E.; Eeles, R.; Shanley, S.; Ardern-Jones, A.; Norman, A.; Kaye, S.B.; Gore, M.E. “BRCAness” syndrome in ovarian cancer: A case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J. Clin. Oncol. 2008, 26, 5530–5536. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Lynch, H.T.; Sanger, W.G.; Pirruccello, S.; Quinn-Laquer, B.; Weisenburger, D.D. Familial multiple myeloma: A family study and review of the literature. J. Natl. Cancer Inst. 2001, 93, 1479–1483. [Google Scholar] [CrossRef]
- Chen, Q.; Van der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005, 106, 698–705. [Google Scholar] [CrossRef]
- Patel, P.R.; Senyuk, V.; Sweiss, K.; Calip, G.; Oh, A.; Mahmud, N.; Rondelli, D. Over-coming Melphalan Resistance by Targeting Crucial DNA Repair Pathways in Multiple Myeloma. Biol. Blood Marrow Transplant. 2020, 26, S224–S225. [Google Scholar] [CrossRef]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A. Alt FW Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000, 404, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Betti, C.; Singh, S.; Toor, A.; Vaughan, A. Impaired NHEJ function in multiple myeloma. Mutat. Res. 2009, 660, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hayden, P.J.; Tewari, P.; Morris, D.W.; Staines, A.; Crowley, D.; Nieters, A.; Becker, N.; de Sanjosé, S.; Foretova, L.; Maynadié, M.; et al. Variation in DNA repair genes XRCC3, XRCC4, XRCC5 and sus-ceptibility to myeloma. Hum. Mol. Genet. 2007, 16, 3117–3127. [Google Scholar] [CrossRef]
- Roddam, P.L.; Rollinson, S.; O'Driscoll, M.; Jeggo, P.A.; Jack, A.; Morgan, G.J. Genetic variants of NHEJ DNA ligase IV can affect the risk of developing multiple myeloma, a tumour characterised by aberrant class switch recombination. J. Med. Genet. 2002, 39, 900–905. [Google Scholar] [CrossRef]
- Roddam, P.L.; Allan, J.M.; Dring, A.M.; Worrillow, L.J.; Davies, F.E.; Morgan, G.J. Non-homologous end-joining gene profiling reveals distinct expression patterns associated with lymphoma and multiple myeloma. Br. J. Haematol. 2010, 149, 258–262. [Google Scholar] [CrossRef]
- Calimeri, T.; Fulciniti, M.; Lin, J.; Samur, M.K.; Calkins, A.S.; Vahia, A.V.; Pal, J.; Cea, M.; Cagnetta, A.; Cottini, F.; et al. Aberrant non-homologous end joining in multiple myeloma: A role in genomic instability and as potential prognostic marker. Blood 2012, 120, 2932. [Google Scholar] [CrossRef]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Duquette, M.L.; Zhu, Q.; Taylor, E.R.; Tsay, A.J.; Shi, L.Z.; Berns, M.W.; McGowan, C.H. CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS Genet. 2012, 8, e1003050. [Google Scholar] [CrossRef]
- Longerich, S.; Li, J.; Xiong, Y.; Sung, P.; Kupfer, G.M. Stress and DNA repair biology of the Fanconi anemia pathway. Blood 2014, 124, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Gourzones-Dmitriev, C.; Sahota, S.; Rème, T.; Moreaux, J.; Goldschmidt, H.; Constantinou, A.; Pasero, P.; Hose, D.; Klein, B. A DNA repair pathway score predicts survival in human multiple myeloma: The potential for therapeutic strategy. Oncotarget 2014, 5, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Bollati, V.; Fabris, S.; Pegoraro, V.; Ronchetti, D.; Mosca, L.; Deliliers, G.L.; Motta, V.; Bertazzi, P.A.; Baccarelli, A.; Neri, A. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis 2009, 30, 1330–1335. [Google Scholar] [CrossRef]
- Sive, J.I.; Feber, A.; Smith, D.; Quinn, J.; Beck, S.; Yong, K. Global hypomethylation in myeloma is associated with poor prognosis. Br. J. Haematol. 2016, 172, 473–475. [Google Scholar] [CrossRef]
- Rizwana, R.; Hahn, P.J. CpG methylation reduces genomic instability. J. Cell Sci. 1999, 112 Pt 24, 4513–4519. [Google Scholar]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Maes, K.; De Smedt, E.; Lemaire, M.; De Raeve, H.; Menu, E.; Van Valckenborgh, E.; McClue, S.; Vanderkerken, K.; De Bruyne, E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014, 5, 3115–3129. [Google Scholar] [CrossRef]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Gobbi, M.; Patrone, F.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. New insights into the treatment of multiple myeloma with histone deacetylase inhibitors. Curr. Pharm. Des. 2013, 19, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, N.C.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; De Las Rivas, J.; Ticona, F.V.; Fermiñán, E.; Martín-Jiménez, P.; Chillón, C.; Risueño, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Favasuli, V.; Ronchetti, D.; Todoerti, K.; Pelizzoni, F.; Manzoni, M.; Barbieri, M.; Fabris, S.; Silvestris, I.; Gallo Cantafio, M.E.; et al. Long non-coding RNA NEAT1 targeting impairs the DNA repair machinery and triggers anti-tumor activity in multiple myeloma. Leukemia 2020, 34, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lin, J.; Fang, H.; Fang, J.; Li, C.; Chen, W.; Liu, S.; Ondrejka, S.; Gong, Z.; Reu, F.; et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia 2018, 32, 2250–2262. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoffc, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer’s disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Hamidzadeh, K.; Christensen, S.M.; Dalby, E.; Chandrasekaran, P.; Mosser, D.M. Macrophages and the Recovery from Acute and Chronic Inflammation. Annu. Rev. Physiol. 2017, 79, 567–592. [Google Scholar] [CrossRef]
- Hou, J.; Wei, R.; Qian, J.; Wang, R.; Fan, Z.; Gu, C.; Yang, Y. The impact of the bone marrow microenvironment on multiple myeloma (Review). Oncol. Rep. 2019, 42, 1272–1282. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Cretu, A.; Brooks, P.C. Impact of the non-cellular tumor microenvironment on metastasis: Potential therapeutic and imaging opportunities. J. Cell Physiol. 2007, 213, 391–402. [Google Scholar] [CrossRef] [PubMed]
- De Beule, N.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E.; Breckpot, K.; De Raeve, H.; Van Rampelbergh, R.; Van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J. Pathol. 2017, 241, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2018, 25, 101084. [Google Scholar] [CrossRef]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef]
- Wu, Z.H.; Shi, Y.; Tibbetts, R.S.; Miyamoto, S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006, 311, 1141–1146. [Google Scholar]
- Caillot, M.; Zylbersztejn, F.; Maitre, E.; Bourgeais, J.; Hérault, O.; Sola, B. ROS Overproduction Sensitises Myeloma Cells to Bortezomib-Induced Apoptosis and Alleviates Tumour Microenvironment-Mediated Cell Resistance. Cells 2020, 9, 2357. [Google Scholar] [CrossRef]
- Bustany, S.; Bourgeais, J.; Tchakarska, G.; Body, S.; Hérault, O.; Gouilleux, F.; Sola, B. Cyclin D1 unbalances the redox status controlling cell adhesion, migration, and drug resistance in myeloma cells. Oncotarget 2016, 7, 45214–45224. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, O.; Gong, G.; Olipitz, W.; Muthupalani, S.; Engelward, B.P. Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet. 2015, 11, e1004901. [Google Scholar] [CrossRef] [PubMed]
- Nerini-Molteni, S.; Ferrarini, M.; Cozza, S.; Caligaris-Cappio, F.; Sitia, R. Redox homeostasis modulates the sensitivity of myeloma cells to bortezomib. Br. J. Haematol. 2008, 141, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Saba, F.; Soleimani, M.; Abroun, S. New role of hypoxia in pathophysiology of multiple myeloma through miR-210. EXCLI J. 2018, 17, 647–662. [Google Scholar]
- Kocemba-Pilarczyk, K.A.; Trojan, S.; Ostrowska, B.; Lasota, M.; Dudzik, P.; Kusior, D.; Kot, M. Influence of metformin on HIF-1 pathway in multiple myeloma. Pharmacol. Rep. 2020, 72, 1407–1417. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, S.E.; Glazer, P.M. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair 2015, 32, 180–189. [Google Scholar] [CrossRef]
- Begg, K.; Tavassoli, M. Inside the hypoxic tumour: Reprogramming of the DDR and radioresistance. Cell Death Discov. 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.; Hill, R. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- El Arfani, C.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E. Metabolic Features of Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1200. [Google Scholar] [CrossRef]
- Nakano, A.; Miki, H.; Nakamura, S.; Harada, T.; Oda, A.; Amou, H.; Fujii, S.; Kagawa, K.; Takeuchi, K.; Ozaki, S.; et al. Up-regulation of hexokinaseII in myeloma cells: Targeting myeloma cells with 3-bromopyruvate. J. Bioenerg. Biomembr. 2012, 44, 31–38. [Google Scholar] [CrossRef]
- Giuliani, N.; Chiu, M.; Bolzoni, M.; Accardi, F.; Bianchi, M.G.; Toscani, D.; Aversa, F.; Bussolati, O. The potential of inhibiting glutamine uptake as a therapeutic target for multiple myeloma. Expert Opin. Ther. Targets 2017, 21, 231–234. [Google Scholar] [CrossRef]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chang, J.F.; Sun, J.; Chen, L.; Yang, X.M.; Tang, H.Y.; Jing, Y.Y.; Kang, X.; He, Z.M.; Wu, J.Y.; et al. Histone H3K27 methylation modulates the dynamics of FANCD2 on chromatin to facilitate NHEJ and genome stability. J. Cell Sci. 2018, 131, jcs215525. [Google Scholar] [CrossRef] [PubMed]
- Moretton, A.; Loizou, J.I. Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers 2020, 12, 2051. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pathway | Gene Name | Prognostic Value | HR (OS) | HR (EFS) |
|---|---|---|---|---|
| NHEJ | NSD2 | BAD | 3.7 | 2.8 |
| NHEJ | RIF1 | BAD | 3.2 | 2.3 |
| NHEJ | XRCC5 | BAD | 2.9 | 2.5 |
| NHEJ | PNKP | GOOD | 0.4 | 0.5 |
| NHEJ | POLL | GOOD | 0.3 | 0.5 |
| HRR/MMR | EXO1 | BAD | 3.9 | 1.8 |
| HRR/MMR | BLM | BAD | 2.9 | 1.8 |
| HRR/NER | RPA3 | BAD | 3.2 | 3.1 |
| HRR | RAD51 | BAD | 2.8 | 1.7 |
| HRR | MRE11 | BAD | 2.1 | 1.8 |
| HRR | ATM | GOOD | 0.5 | 0.6 |
| FA | RMI1 | BAD | 5 | 3 |
| FA | FANCI | BAD | 3.5 | 2.4 |
| FA | FANCA | BAD | 2.2 | 2.4 |
| NER | PCNA | BAD | 4.5 | 2.2 |
| NER/HRR | RPA3 | BAD | 3.2 | 3.1 |
| NER/BER/a-NHEJ | LIG3 | BAD | 2.6 | 2 |
| NER | POLD3 | BAD | 6.3 | 2.1 |
| NER | ERCC4 | BAD | 2.5 | 2.2 |
| NER | POLD1 | BAD | 2.4 | 2 |
| NER | ERCC1 | GOOD | 0.4 | 0.4 |
| NER | ERCC5 | GOOD | 0.5 | 0.5 |
| MMR/HRR | EXO1 | BAD | 3.9 | 1.8 |
| MMR | MSH2 | BAD | 2.7 | 1.6 |
| BER/NER/a-NHEJ | LIG3 | BAD | 2.6 | 2 |
| Genes | DDR Pathway | Expression | OS | Drug Resistance | Inhibitors | References | |
|---|---|---|---|---|---|---|---|
| APE1 | BER (HR) | Increased | Poor | Yes | API3 | Sensitize Mel | PMID: 28938675 |
| APE2 | BER | Increased | Poor | PMID: 28938675 | |||
| PARP1 | BER, Alt-NHEJ | Increased | Poor | Yes | Olaparib, PJ34 | Sensitize Mel | PMID: 24928009, PMID: 32079692 |
| ERCC3 | NER | Increased | Poor | Yes | Spironolactone, Triptolide | Sensitize Mel | PMID: 28588253 |
| RAD51 | HR | Increased | Poor | Yes | B02 | Sensitize Mel | PMID: 25996477 |
| DCLRE1C | c-NHEJ | Increased | Poor | PMID: 23966156 | |||
| XRCC5 | c-NHEJ | Increased | Poor | PMID: 23966156 | |||
| LIG3 | BER, NER, alt-NHEJ | Increased | Poor | Yes | miR-22 | Sensitize Bor | PMID: 30120376 |
| ATR | Signaling | VX-970 | Sensitize Mel | PMID: 33054085 | |||
| CHEK2 | Effector | Dinaciclib | Sensitize PARP | PMID: 26719576 | |||
| DNMT1 | DNA methyltransferase | Increased | Decitabine | Sensitize HR | PMID: 24833108 | ||
| HDAC1 | Histone deacetylase | Increased | Poor | Yes | Panobinostat | Sensitize Bor | PMID: 32267687 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saitoh, T.; Oda, T. DNA Damage Response in Multiple Myeloma: The Role of the Tumor Microenvironment. Cancers 2021, 13, 504. https://doi.org/10.3390/cancers13030504
Saitoh T, Oda T. DNA Damage Response in Multiple Myeloma: The Role of the Tumor Microenvironment. Cancers. 2021; 13(3):504. https://doi.org/10.3390/cancers13030504
Chicago/Turabian StyleSaitoh, Takayuki, and Tsukasa Oda. 2021. "DNA Damage Response in Multiple Myeloma: The Role of the Tumor Microenvironment" Cancers 13, no. 3: 504. https://doi.org/10.3390/cancers13030504
APA StyleSaitoh, T., & Oda, T. (2021). DNA Damage Response in Multiple Myeloma: The Role of the Tumor Microenvironment. Cancers, 13(3), 504. https://doi.org/10.3390/cancers13030504