1. Introduction
The ‘liquid biopsy’ analysis of the peripheral blood of cancer patients has made remarkable progress [
1]. The subjects of liquid biopsy are circulating tumor cells (CTCs) or molecules like nucleic acids or proteins that are released by tumor cells into the blood. When such tumor-derived biomaterial is detected in the blood, it can be used for the early detection of cancer, the prediction of metastatic relapse, or therapy monitoring [
2].
In breast cancer, the detection of CTCs in the blood, and their relatives that have already reached secondary sites—the disseminated tumor cells in the bone marrow (DTCs)—are useful markers for the prediction of distant metastasis [
3]. DTCs survive chemotherapy and predict poor clinical outcomes of the patients [
4,
5,
6,
7]. However, there is still a substantial number of patients who relapse despite negative blood (CTCs) or bone marrow (DTCs) findings at their primary diagnosis [
5,
8,
9], reflecting the heterogeneity of the disseminating tumor cells [
3]. However, most studies have employed antibodies against epithelial cell markers in order to detect CTCs or DTCs [
10]. Besides breast carcinoma with an epithelial phenotype, there is also a fluent transition of phenotypes with varying degrees of mesenchymal attributes [
11,
12,
13]. Hence, due to a reduced moulding of epithelial characteristics, DTCs and CTCs with the pronounced manifestation of mesenchymal attributes and a weak expression of epithelial markers (mDTC and mCTC) are difficult to detect by epithelial cell markers. Recent experiments have confirmed the presence of such CTCs in breast [
14] and lung cancer patients [
15]. Thus, there is an urgent need for new markers that support the detection of mDTCs and mCTCs in routine analysis.
One factor that can promote the acquisition of mesenchymal attributes and the dissemination of tumor cells is hypoxia [
16,
17]. Hypoxia can be present in the primary tumor and in secondary metastatic sites like the bone marrow, where concentrations of only 1% O
2 (hypoxia) are detected [
18,
19]. We previously detected microenvironmental stress-tolerant mDTCs in the bone marrow that were strongly positive for proteins of hypoxic stress-response programs [
20]. Hence, the proteins of stress response programs could be applicable as markers for the detection of mCTCs and mDTCs that are difficult to identify by the established markers used in previous studies.
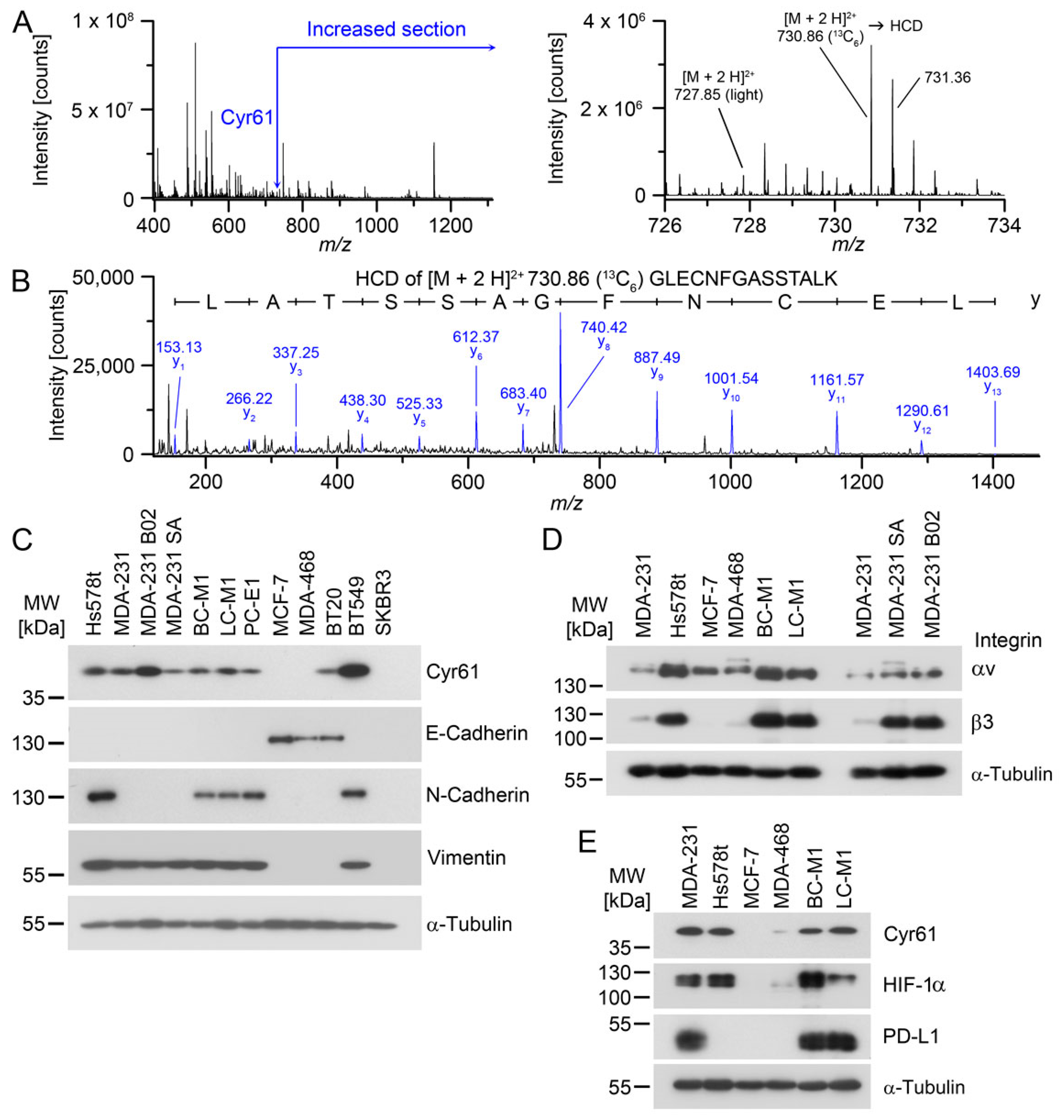
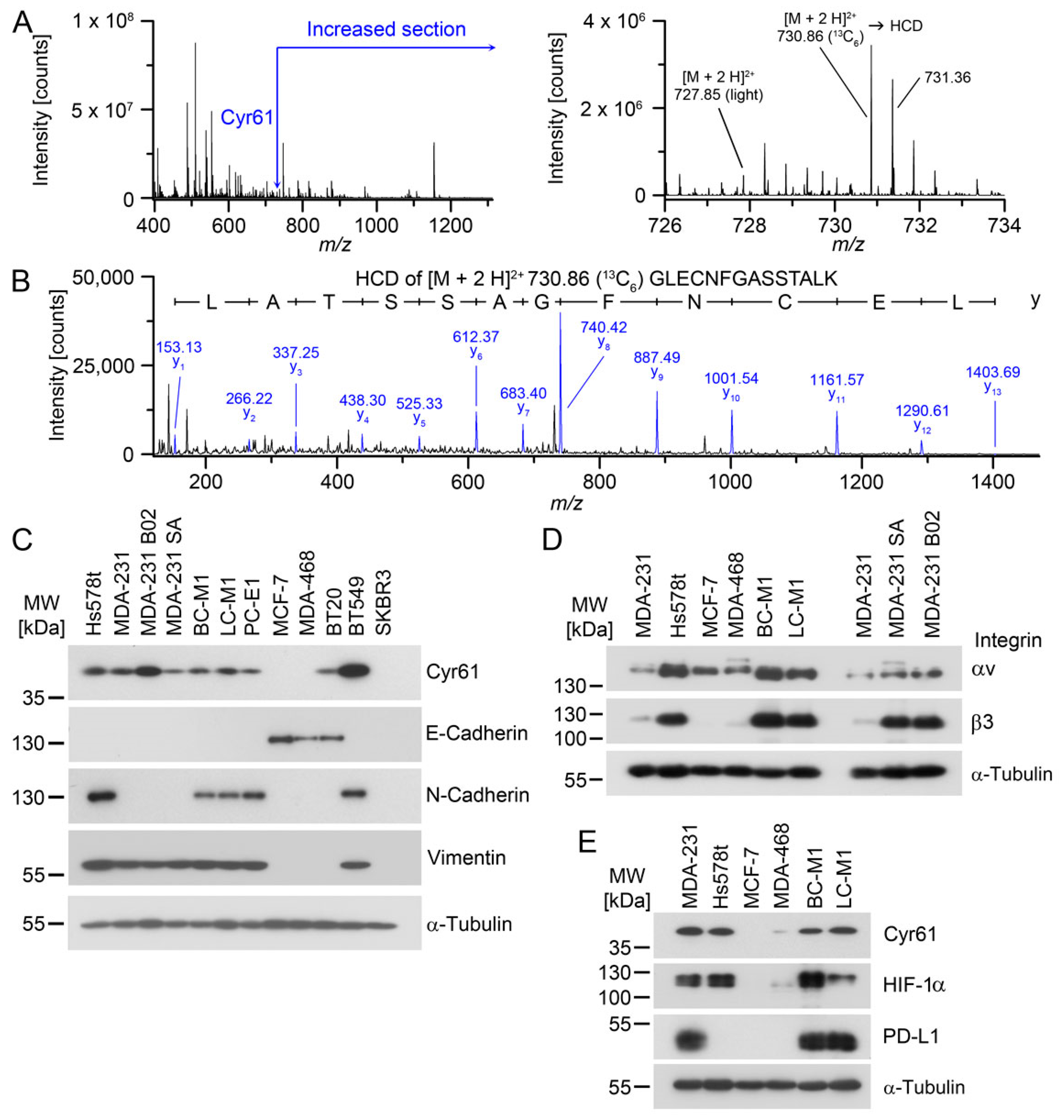
The 42 kDa protein cysteine-rich angiogenic inducer 61 (Cyr61) is a versatile responder for microenvironmental stress, and can be secreted to the extracellular space. Cyr61 has been implicated in migration, cellular differentiation, and the induction of angiogenesis [
21]. In breast cancer, about thirty percent of invasive tumors show elevated Cyr61 expression compared to normal tissue, and higher levels of Cyr61 are associated with the formation of metastasis [
22,
23]. The promotion of metastasis by Cyr61 is mediated by enabling tumor cell extravasation and protecting tumor cells from anoikis during the dissemination process [
24]. However, the detailed analysis of the pathophysiologic role of Cyr61 is complicated by its complex and dynamic regulation, leading to diverse and sometimes opposing cellular responses depending on the individual cell context [
25].
Three major factors were discovered that influence the expression of Cyr61. First, the stimulation of the epidermal growth factor receptor (EGFR) can induce an increase in cytoplasmic Cyr61 levels [
26], while the effect on secreted Cyr61 was not investigated. Another function of Cyr61 which is relevant in metastasis is that Cyr61 is a hypoxia-inducible angiogenesis factor which might interact with hypoxia-inducible factor-1-alpha (HIF-1α) as a master regulator of the metabolic adaptation to hypoxia. This effect might support the idea of a hypoxia-induced selection process during tumor progression towards tumor cells with elevated Cyr61 expression [
27]. Secreted Cyr61 can act as a ligand for different integrin species, of which the integrin αv/integrin β3 heterodimer appears to be the favoured receptor for Cyr61 [
22].
We have previously observed that breast cancer cells with low keratin expression due to an epithelial–mesenchymal transition show elevated levels of the immune checkpoint regulator programmed death-ligand 1 (PD-L1), and that PD-L1 is induced by hypoxia in such cells [
28]. Therapies that are directed against the control of the immune checkpoints have become interesting novel approaches, and PD-L1 is currently one of the most prominent therapeutic targets [
29]. PD-L1 has been detected in CTCs using keratins as CTC-detection proteins [
30]. However, it might be possible that additional breast cancer cells that only weakly express keratins display high levels of Cyr61 and PD-L1, and are therefore overlooked by standard CTC assays. In particular, the remarkably-quick response of PD-L1 to changing oxygen concentrations [
28] attracted our interest here to study a potential link between Cyr61 and PD-L1. To our best knowledge, no reports have been published on Cyr61 expression in the CTCs or DTCs of cancer patients.
The principal goal of this study was to determine whether Cyr61 may constitute a robust marker for CTC/DTC, and we therefore aimed to examine the fluctuations of Cyr61 levels in various tumor cell lines and DTC cell lines cultivated in different conditions that mimic various microenvironmental stresses encountered by CTCs and DTCs. First, we used a mass-spectrometry–based approach that revealed the protein Cyr61 in mDTC cell lines. We then investigated which common biological parameters that are relevant to breast cancer progression (e.g., ErbB-2, microenvironmental stress) affect the cytoplasmic levels of Cyr61 in tumor cells. These results provide important information on the expected range of Cyr61 levels in CTC or DTC in clinical samples from the breast cancer patients analysed in this investigation.
3. Discussion
Even though recent developments in CTC research have provided new insights into cancer biology, and have opened novel areas of cancer diagnostics, new markers for the detection and characterization of CTCs are still needed [
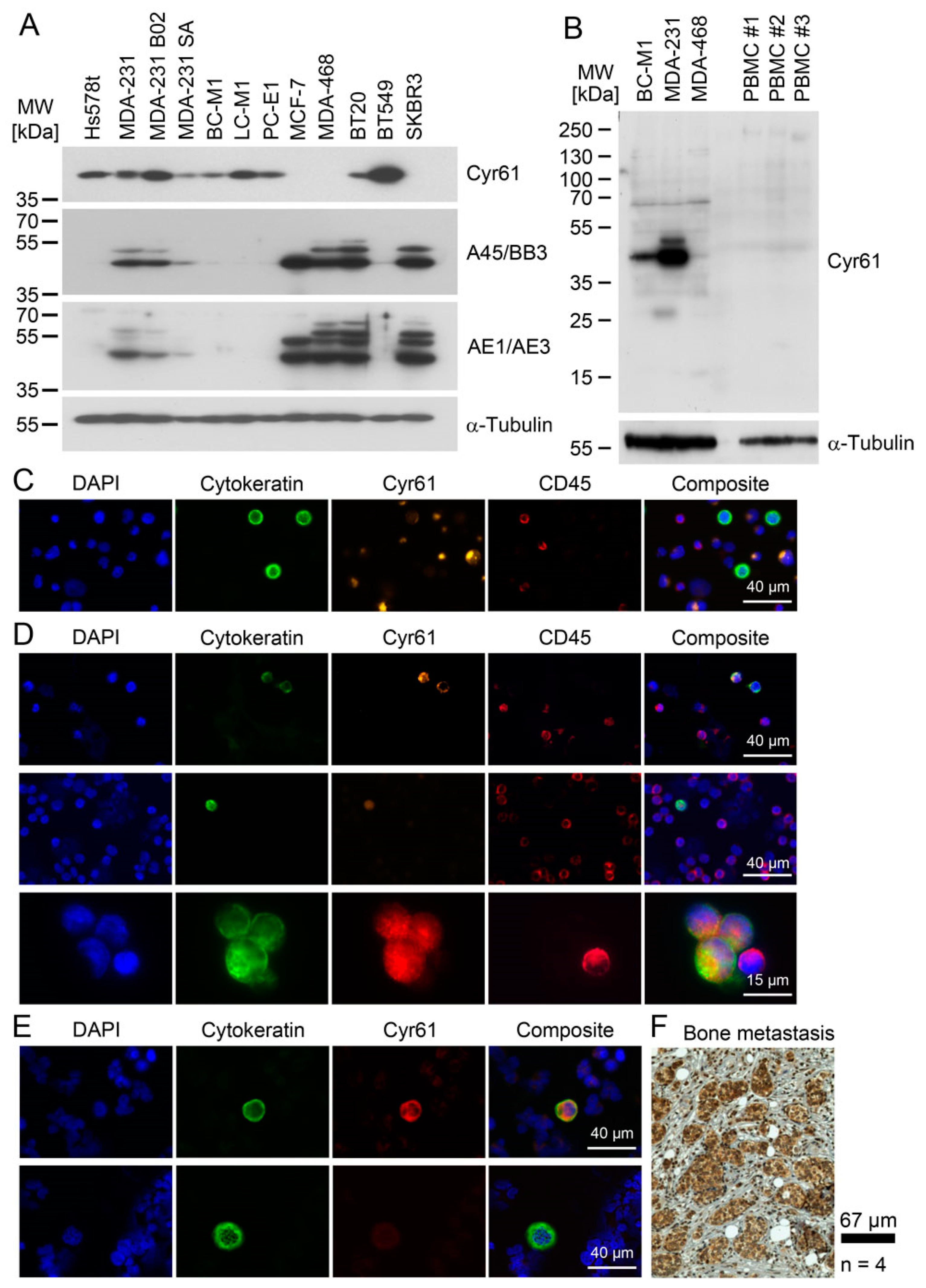
1]. Here, we introduced the stress-related protein Cyr61 as a potential new marker for CTCs. In order to detect Cyr61 in CTCs, sensitive multiplex immunostaining assays were developed, which allowed us to detect even breast tumor cells with low keratin levels. CTCs that weakly express keratins (as the standard CTC marker) are difficult to assess against background fluorescence; if these cells show a strong Cyr61 staining, it may help to reduce false negative results. We observed a marked heterogeneity of Cyr61 levels in CTCs both with regard to inter- and intra-patient variability, suggesting that CTCs have different abilities to respond to environmental stress.
We modelled, in cell lines, the response of Cyr61 to the pathophysiological parameters typically found in breast cancer patients in order to judge the usefulness of Cyr61 as a CTC/DTC detection marker in practice. These experiments revealed that the levels of Cyr61 are quite variable, depending on the specific stimulus, yet the overall Cyr61 expression is sufficient high in mCTC/mDTC for a confident detection in CTC/DTC from cancer patients.
For cytokeratin detection in CTC/DTC, we applied a sensitive and broad-range pan-cytokeratin antibody cocktail. We previously detected CK5 in the DTC cell lines on a low level by Western blot analysis [
20,
41]; therefore, we applied the keratin-specific antibody AE1/AE3 (together with the antibody C11), which detects CK5 with high sensitivity [
42]. By this approach, we could detect both cancer cells with a strongly epithelial phenotype and cancer cells that show only a very low degree of cellular differentiation with low keratin expression [
43]. This helped us to confirm the presence of Cyr61 in CTC from cancer patients, as our detection approach was suited to a CK5/Cyr61 positive phenotype like BC-M1, but could also detect CTCs with a high degree of epithelial differentiation. Interestingly, no Cyr61-positive/cytokeratin-negative cells were found, which might be an effect of our analytical approach emphasizing the detection of a tumor cell phenotype like BC-M1. Alternatively, breast cancer cells with a complete lack of all cytokeratin proteins might be very rare, or do not express Cyr61 [
44].
Keratins are present in the cells as heteropolymers [
45], which are difficult to disrupt to monomers by normal protein extraction methods. For the conversion to the keratin monomers that are then analysable by SDS-PAGE, the application of high-molar urea solutions is a suitable approach [
46]. However, it cannot be excluded that the disruption to monomers is not complete, leading to a partial precipitation of keratin polymers during the SDS-PAGE. This might explain the low keratin signals for the DTC cell lines in some of our Western blots, even though we confirmed—by 2-D electrophoresis and mass spectrometry—that BC-M1 displays a reduced keratin expression pattern [
41]. Therefore, a certain discrepancy in the measured keratin levels of the cell lines compared with those on CTCs of cancer patients might be due to keratin detection on CTCs by immunocytochemical detection, whereas for the cell lines, Western blot analysis was used.
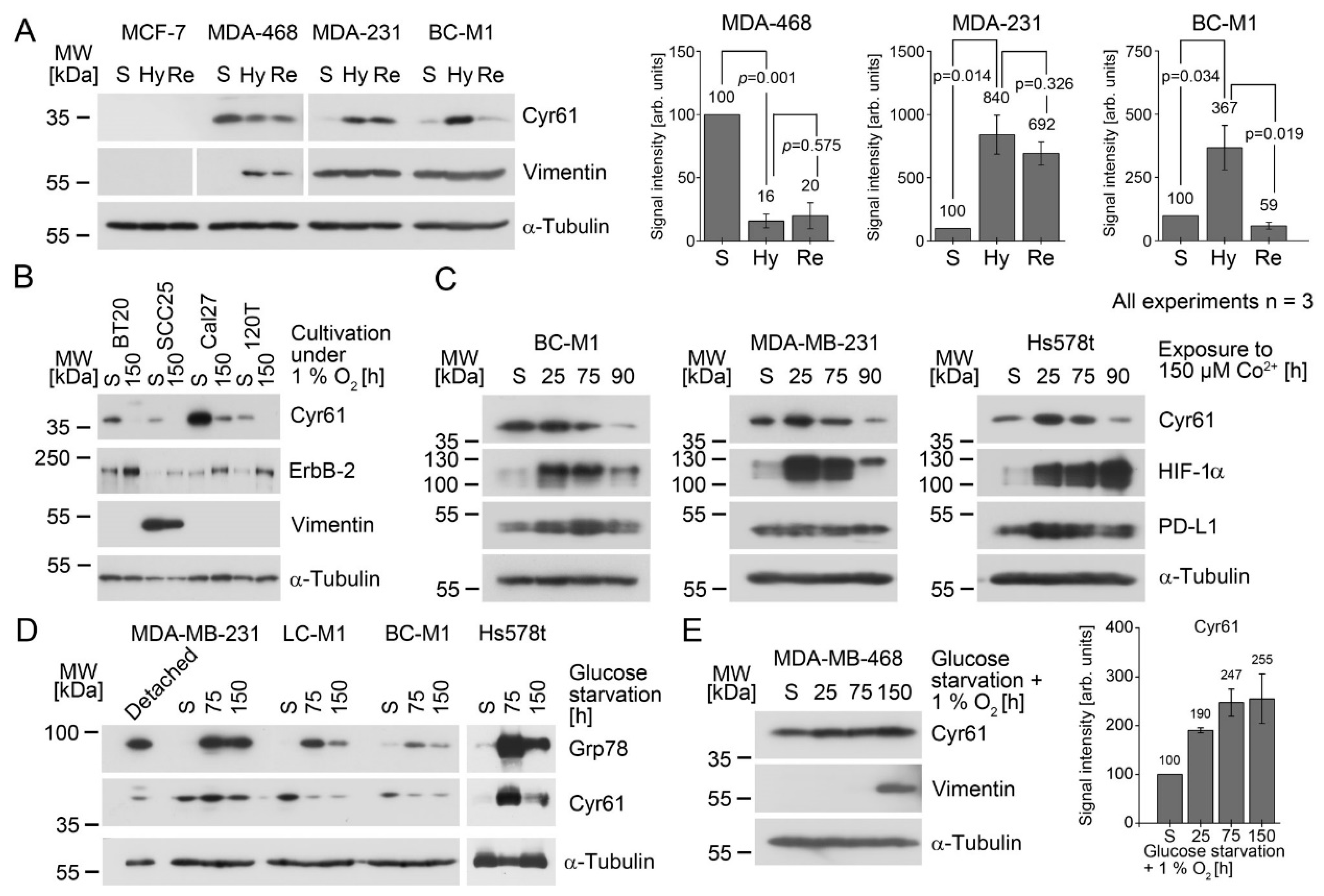
We further interrogated the involvement of Cyr61 in stress responses that were relevant to disseminating tumor cells. After extravasation, CTCs may encounter a hypoxic microenvironment (e.g., in bone marrow), which affects the protein expression responding to this stress condition [
47,
48]. We therefore subjected the cell lines to 1% O
2, which is the lower limit of the oxygen concentration in the bone marrow [
19] and is sufficient to stabilize HIF-1α in most human cells [
49]. In melanoma cells, a regulation of Cyr61 by HIF-1α was suggested, which is not mediated by the direct binding of HIF-1α to the Cyr61 promoter, but via c-Jun/AP-1 [
27]. As it was also shown that high Cyr61 levels are associated with a more-aggressive breast cancer phenotype, this supports the idea of a selection process in vivo, which finally leads to aggressive phenotypes with high Cyr61 expression [
27]. Our findings here support this idea, as all three investigated DTC cell lines were positive for Cyr61, and the bone metastasis sublines of MDA-MB-231—B02 and SA—also displayed high Cyr61 levels. Furthermore, we detected Cyr61 in the CTCs and overt bone metastases of breast cancer patients, closing the gap between the cell line experiments and the in situ findings in clinical specimens.
On the other hand, there might be an antagonistic regulative pathway that suppresses the Cyr61 levels under hypoxia. One candidate might be ErbB-2, which is strongly induced under hypoxia in MDA-MB-468 [
28]. We observed here a downregulation of Cyr61 in this cell line. Cyr61 knock down did not affect the ErbB-2 levels in MDA-MB-231, suggesting that the ErbB-2 affects Cyr61 levels, but not vice versa.
Cyr61 is a matricellular protein present in both the cytoplasm and extracellular space [
25]. In the extracellular region, it can act as a ligand for numerous receptors like the integrins. Unlike previous analyses by others, we simultaneously analyzed both the cytoplasmic and the secreted Cyr61 protein levels in order to obtain deeper insights into the biology of Cyr61. Moreover, the presence of cytoplasmic Cyr61 is crucial for the detection of CTC and DTC by Cyr61. Therefore, we investigated the possibility that tumor cells secrete large amounts of Cyr61 while maintaining cytoplasmic Cyr61 levels facilitating CTC and DTC detection even after secretion of large amounts of Cyr61 into the extracellular space.
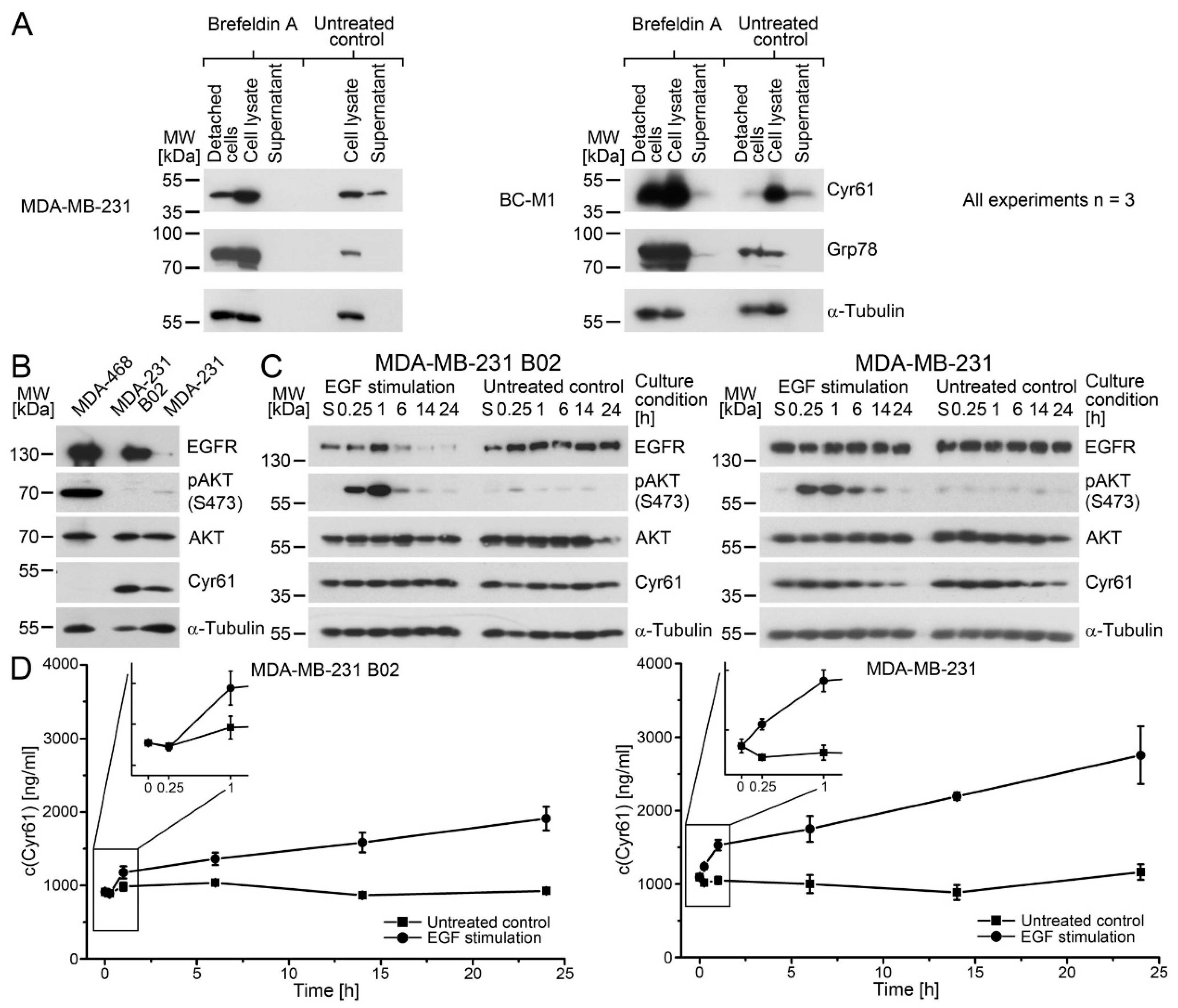
As it has been reported before for endometrial cells that the
cyr61 mRNA can be induced by EGF stimulation via EGFR activation [
26], we selected the EGFR-positive cell line MDA-MB-231 as a model. The activation of AKT after EGF treatment showed that the cells responded to the EGF stimulation, whereas the Cyr61 levels in the cytoplasm remained constant. In contrast, we observed a rapid increase in the secreted Cyr61 in the cell culture medium. Our initial hypothesis was that cytoplasmic Cyr61 serves as storages that can be quickly emptied upon the stimulation of the cells leading to Cyr61–weakly-positive cells that are difficult to detect in the circulation. Instead, we observed that the cytoplasmic Cyr61 levels remained constant, further suggesting that the increased total amount in the experiments (cytoplasmic + secreted Cyr61) results from the rapid novel synthesis of the Cyr61 proteins. The rapid induction of Cyr61 proteins might be due to its membership of the immediate-early genes that can be transcribed within minutes after activation [
50]. From that point, a more complete view of the biological function of Cyr61 can be obtained when the secreted Cyr61 is analyzed, in particular, in experiments where no changes in the cytoplasmic levels were detected before.
We observed a similar cellular response of Cyr61 in the B02 cells, even though the EGFR levels in B02 were strongly elevated compared with MDA-MB-231, suggesting that the induction of Cyr61 secretion is not dependent on different EGFR levels in the cells. An interesting effect, which awaits future investigation, was the delayed Cyr61 secretion in B02 compared with MDA-MB-231. A similar effect was observed when we exchanged the cell culture medium for MDA-MB-231 and BC-M1. Future experiments might elucidate the reason for the different Cyr61 secretion kinetics between MDA-MB-231 and the bone marrow resident cells B02 and BC-M1. Finally, these experiments might imply that the amount of secreted Cyr61 is not necessarily proportional to its cytoplasmic form. This idea might be relevant for the interpretation of the Cyr61 levels that were obtained from solid tissue specimens [
22,
51].
Moreover, Cyr61 shows a complex multi-layered regulation, which may have implications for the interpretation of clinical data. In prostate cancer, patients showed a correlation of low Cyr61 protein levels with high recurrence rates [
52]. Others reported, for breast cancer, that
cyr61 mRNA is frequently induced under hypoxia [
53], whereas we found that the cytoplasmic Cyr61 protein is sometimes downregulated. The abrogation of the global mRNA translation under cell stress like hypoxia leads to mRNA accumulation, and to the blockading of the protein synthesis [
47]. For
cyr61, internal ribosome entry sites were detected, as well as the ability of
cyr61-mRNA to undergo low cap-independent protein translation, allowing the synthesis of Cyr61 protein under cell stress [
48,
54,
55]. Furthermore, alternative splice variants of the
cyr61 mRNA were detected in primary breast specimens that displayed the same level as the normal tissue [
53]. Therefore, the correlation of a clinicopathological parameter or a biological response may depend on whether
cyr61-mRNA, or cytoplasmic or secreted Cyr61 protein is being analysed, which may also explain the inconsistencies between published data on Cyr61.
One novel important aspect in cancer therapy is the use of immune checkpoint proteins. As PD-L1 was also detected in CTCs, it is an attractive therapeutic approach to target PD-L1 on CTCs [
29,
30]. Moreover, we previously found a manifest intra-patient and inter-patient heterogeneity in the PD-L1 expression in CTCs from patients with breast cancer [
30]. Epithelial marker proteins like keratins are frequently used for the detection of PD-L1 in CTCs [
30,
56]. Here, we observed that mDTC lines with low keratin expression and mesenchymal attributes are strongly positive for PD-L1 and Cyr61. As Cyr61 is important for the induction of angiogenesis [
57], such Cyr61/PD-L1 double-positive DTC may display a phenotype that is able to escape the immune system, and is able to establish novel tumor cell colonies at secondary sites. This hypothesis might explain the previous findings of other groups that Cyr61 is associated with a more-aggressive phenotype, and is important for metastatic progression in breast cancer [
24]. Furthermore, it was reported that Cyr61 mediates resistance against estrogen [
58], which might hamper endocrine therapies to prevent metastatic breast cancer.
Another regulative layer that affects the Cyr61 levels is the different half-life of the protein in the cytoplasm and the extracellular matrix. For the cytoplasmic Cyr61, a half-life of approximately 20–30 min, and for the extracellular Cyr61, a half-life of more than 24 h has been reported [
50,
59], which might have implications for its application as a CTC detection protein. CTC may experience a re-oxygenation pulse once the tumor cells are released from hypoxic tissue areas into the well-oxygenated blood circulation [
28]. We observed that cytoplasmic Cyr61 is induced in BC-M1 under hypoxia, whereas the Cyr61 levels decreased to approximately 16% after 4 h of the reoxygenation of the cells, suggesting that Cyr61 is quickly degraded upon the reoxygenation of BC-M1. These properties can explain the rapid changes in Cyr61 protein concentrations following the in vitro re-oxygenation model used in his work to mimic the release to CTCs into the blood stream. Again, this effect appears to occur in a cell-line–specific manner, as we did not observe rapidly decreased Cyr61 levels in MDA-MB-231 and MDA-MB-468 after their reoxygenation.
As a consequence, DTC that re-enter the circulation may reduce their cytoplasmic Cyr61 levels during their passage to another secondary site. As the Cyr61 levels in BC-M1 are sufficiently high for confident immunocytochemical analysis, it might be conceivable that the subcellular location of Cyr61 changes during reoxygenation from nucleus-associated to proteasome-associated. If this is true, it would allow the discrimination of freshly-disseminated cells from those that have already spent more time in the circulation.
Recently, we showed that other proteins in CTCs that are therapeutic targets or biomarker proteins (e.g., PD-L1) alsoresponded rapidly to re-oxygenation [
28]. This might be an important aspect to the consideration of whether these proteins are assessed in CTCs in clinical studies as surrogates of the molecular characteristics of tumor cells in primary or metastatic tissues. We previously observed in MDA-MB-231 and BC-M1 for PD-L1 a similar response to re-oxygenation as for Cyr61 in this work, suggesting a common regulative mechanism of these proteins.
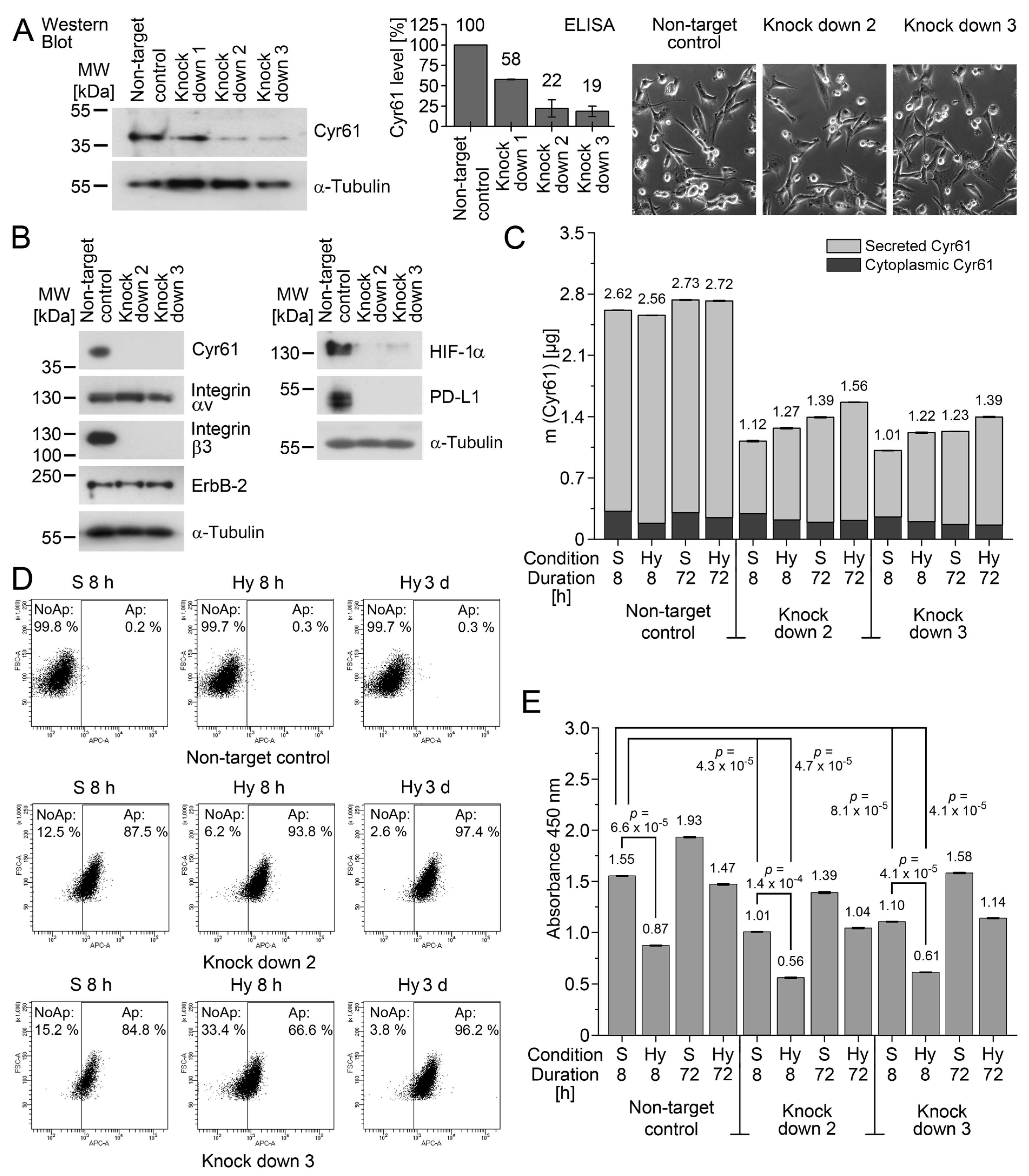
Furthermore, in the Cyr61 knockdown cells, HIF-1α was downregulated, suggesting a regulative axis between Cyr61, c-Jun/AP-1, and HIF-1α, which needs to be further explored in future investigations. We found here that the loss of Cyr61 increases the apoptosis rates of cancer cells, which might be an interesting aspect in the context of PD-L1 detection and targeting in CTCs. Thus, future studies will show whether the Cyr61/PD-L1 double-positive phenotype might characterize a tumor cell population that might be of particular importance in breast cancer metastasis and its response to immunotherapy.
Taken together, Cyr61 might be suitable as a stress-related reporter for very rapid microenvironmental changes. Moreover, Cyr61 might serve as a novel marker for CTCs and DTCs with high plasticity in breast cancer. The remarkable regulation of Cyr61 might enable a perceptive readout of pathophysiological states, which may also provide future information on how to target disseminating cancer cells.
4. Materials and Methods
All of the cell line experiments were performed in biological replicates (nbiol).
4.1. Patients
The human investigations were performed according to the Helsinki rules after approval was obtained by the ethics committee of the Medical Association of Hamburg (reference number PV5392). Written informed consent was obtained from all of the patients prior to any study-related procedures. Samples from women with breast cancer or healthy control persons treated at the University Medical Centre Hamburg-Eppendorf, Germany, were used. The blood from healthy persons was received from the Institute for Transfusion Medicine, University Medical Center Hamburg-Eppendorf. The fresh clinical samples from the breast cancer patients were drawn from breast cancer patients who were positive for distant metastases.
4.2. Cell Lines and Culture Conditions (Standard Cell Culture Condition)
The cultivation of the DTC cell lines was essentially performed as described [
32]. A detailed overview of the generation, authentication, and properties of the DTC cell lines BC-M1 (obtained from the bone marrow of a breast cancer patient), LC-M1 (obtained from the bone marrow of a lung cancer patient), and PC-E1 (obtained from the bone marrow of a prostate cancer patient) were reported before [
20]. The DTC cell lines were cultured at 37 °C in a humidified environment with 5% of carbon dioxide and 10% of oxygen. The oxygen concentration was adjusted by N
2. The culture medium was RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 10 mg/L insulin, 5.5 mg/L transferrin (all from Life Technologies, Darmstadt, Germany), 50 µg/L EGF (Miltenyi Biotec, Bergisch Gladbach, Germany) and 10 µg/L human basic fibroblast growth factor (b-FGF, Miltenyi Biotec). The bone metastatic sublines of MDA-MB-231, MDA-MB-231 SA [
60], and MDA-MB-231 B02 [
61] were cultivated in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal calf serum (FCS) and 2 mM L-glutamine.
The cell lines by kind provision were: MDA-MB-231 B02 (Philippe Clézardin), MDA-MB-231 SA (Theresa A. Guise), Hs578t (Thomas Dittmar). MCF-7 (from ATCC, 2005; ATCC Cat# HTB-22), MDA-MB-231, MDA-MB-468 (DSMZ, Braunschweig, Germany, 09/2016; DSMZ Cat# ACC-738), BT20 (Cell Lines Service, Eppelheim, Germany, 2007; CLS Cat# 300130/p656_BT-20), BT549 (Cell Lines Service, 07/2008; CLS Cat# 300132/p770_BT-549), Hs578t, SCC25, Cal27, SKRBR3; they were cultivated in DMEM with 10% FCS and 2 mM L-glutamine (all from Life Technologies). The authenticated cell lines (last test) were MCF-7 (09/2017), MDA-MB-231 (02/2014), and Hs578t (09/2015). The authentication was performed by Multiplexion, Heidelberg, Germany by multiplex cell authentication (SNP-Profiling).
All of the cell lines were cultured at 37 °C in a humidified environment. The cell lines that were cultivated in RPMI were kept in the presence of 5% of CO2, and the cell lines that were cultured in DMEM were kept in the presence of 10% CO2. With the exception of the DTC cell lines, the remaining gas mixture was atmospheric air. These cell culture conditions are referred as to ‘standard cell culture conditions’ in this work. The cell lines were stored as cryo-cultures in liquid nitrogen, and the cells were temporarily resuscitated only for the experiments. After resuscitation, the cells were routinely tested for mycoplasmas using the VenorGeM Classic mycoplasma detection kit (Minerva Biolabs, Berlin, Germany, Cat. No. 11-1100). The mycoplasma test was performed after five passages (i.e., ±14 days), followed by the generation of fresh cyro-cultures if the cells were negative for mycoplasma. The mycoplasma-infected cells were expunged and the incubator was subjected to disinfestation. The experiments were performed from passage six to a maximum of 15 passages. The protein samples were generated within six months after the resuscitation of the cell lines.
4.3. Stable Isotope Labelling with Amino Acids in Cell Culture (SILAC) and Mass Spectrometry
The analysis of the protein expression profile of the breast cancer cell lines MDA-MB-468 and BC-M1 was performed as described [
32], with the following modifications. The LC-MS/MS measurements were performed by injecting the samples onto a nano liquid chromatography system (Dionex UltiMate 3000 RSLCnano, Thermo Scientific, Bremen, Germany) coupled via electrospray-ionization (ESI) to a linear trap quadrupole (LTQ) orbitrap mass spectrometer (Orbitrap Fusion, Thermo Scientific, Bremen, Germany). The samples were loaded (3 µL/min) onto a trapping column (Acclaim PepMap nano trap column, C18, 3 µm, 150 mm × 0.075 mm, 100 Ǻ, Thermo Scientific, Bremen, Germany, buffer A: 0.1% FA in HPLC-H2O; buffer B: 0.1% FA in ACN) with 2% buffer B. After the sample loading, the trapping column was washed for 5 min with 2% buffer B (3 μL/min). The peptides were eluted (300 nL/min) onto the separation column (Acclaim PepMap 100, C18, 75 μm × 500 mm, 2 µm, 100 Ǻ, Thermo Scientific, Bremen, Germany) with a gradient of 2−30% B in 30 min. The spray was generated from a fused-silica emitter (I.D. 10 μm, New Objective, Woburn, MA, USA) at a capillary voltage of 1800 V. The mass spectrometric analysis was performed in the positive ion mode. The MS/MS analysis was carried out in data dependent acquisition mode (DDA) in the top speed mode using the following parameters: an HCD collision energy of 28%, an ion intensity threshold of 1 × 10
4, and a quadrupole isolation width of 1.6
m/
z. Every second, an MS scan was performed over an m/z range from 400–1500, with a resolution of 120,000 full width at half maximum (FWHM) at
m/
z 200 (transient length = 256 ms, maximum injection time = 50 ms, AGC target = 2 × 10
5). The MS/MS spectra were recorded in the ion trap (scan-rate = 66 kDa/s, maximum injection time = 200 ms, AGC target = 1 × 10
4). For the peptide and protein identification, Proteome Discoverer 2.0 with Sequest HT (Thermo Scientific, Bremen, Germany) and MaxQuant with Andromeda (version 1.5.2.8) was used. The MS/MS spectra were searched against a human SwissProt database (
www.uniprot.org, downloaded 17 November 2017, 20,239 entries) and a contaminant database (298 entries). The searches were performed using the following parameters: the precursor mass tolerance was set to 10 ppm, and the fragment mass tolerance was set to 0.5 Da. Furthermore, two missed cleavages were allowed, as well as a carbamidomethylation on the cysteine residues as a fixed modification, an oxidation of the methionine residues, and a
13C
6-label of the lysine and arginine residues as a variable modification. The peptides were identified with an FDR of 1%. The proteins were kept as correctly identified when at least one unique peptide was identified.
4.4. Special Cell Culture Conditions
Under special cell culture conditions, the culture conditions were the same as for the standard cell culture conditions, with the following modifications for each experiment. The cultivation of the cell lines in the presence of 1% O
2 (hypoxia) was performed using the incubator Heracell 15 (Thermo Fisher Scientific, Waltham, MA, USA). The oxygen partial pressure was adjusted using N
2. For the reoxygenation experiments, the cells were cultured under hypoxia for 14 days, followed by a reoxygenation pulse of 10% O
2 for 4 h, which mimics the situation of CTCs in the blood stream. Ten percent oxygen is the average value for larger blood vessels, and the duration of 4 h is comparable to the life span of CTC in the blood circuit [
35,
36].
When the cell lines were cultured in a medium that contained no glucose (Glu
0), the media “DMEM, no glucose” and “RPMI, no glucose” (both Life Technologies) were used. For the glucose starvation experiments in presence of 1% O
2, both conditions were combined. For stabilization of HIF-1α, the cells were incubated with 150 µM cobalt chloride (Sigma-Aldrich, Munich, Germany) [
62]. For the Brefeldin A (BFA) treatment, BFA was applied in order to analyse the Cyr61 secretion in cultured cells. The BFA was purchased from Merck (Calbiochem, Darmstadt, Germany, Cat. No. 203729-1MG), and was dissolved in DMSO in a concentration of 10 mg/mL (stock solution). The stock solution was diluted in 12 mL cell culture medium without FCS to a final concentration of 5 µg/mL for each 75 cm
2 cell culture flask. The cells were incubated with BFA for 18 min under the standard cell culture conditions. For the control cells, DMSO without BFA was applied. The centrifugation steps were performed at 0 °C until the proteins of the culture medium were not dissolved in the lysis buffer (9.8 M urea, 15 mM EDTA, 30 mM Tris). The culture medium was collected and centrifuged at 2000×
g for 5 min. The cell pellet of the detached cells was washed with 8 mL PBS, and the cells were lysed with lysis buffer and processed as described for the cell harvest for Western blot analysis. The supernatant (12 mL per cell culture flask) was concentrated by ultraspin centrifugal devices (Vivaspin 4, 10,000 Da MWCO, PES membrane; Sartorius-Stedim, Göttingen, Germany) to a volume of 200 µL. After addition of 3 mL lysis mix the supernatant was concentrated again to a volume of 200 µL. The cell pellet was harvested as described for the cell harvest for Western blot analysis. The three fractions (detached cells, cell lysate and supernatant) were subjected to Western blot analysis, as described. For the BFA experiments 50 µg of protein were applied for Western blot analysis. As BFA treatment leads to the activation of Grp78 [
39], Grp78 induction served as a positive control. As the total protein amount for two of the three biological replicates of the MDA-MB-231 detached cells (untreated) was below 50 µg, a Western blot analysis lacking this sample is shown.
The EGF stimulation was performed as described [
20], with the following modifications. For the EGF stimulation of MDA-231 and MDA-231 B02, the cells were cultured for 48 h in order to enrich the culture medium with secreted Cyr61 from the cells. Then, the culture medium was supplemented with 100 ng EGF per milliliter and incubated for the appropriate duration of time. As controls, cells were cultured without the addition of EGF to the culture medium in parallel. The cell culture supernatant was harvested, clarified by centrifugation, and analyzed for Cyr61 by an enzyme-linked immunosorbent assay, as described below.
4.5. Cell Harvest and Sample Procurement for Western Blot
The cells from the cell lines were washed three times with 37 °C prewarmed PBS and harvested in 300 µL lysis mix (9.8 M urea, 15 mM EDTA, 30 mM Tris) per 75 cm2 cell culture flask. The cell lysates were homogenized on ice by ultrasonic treatment using the ultrasonic device UP50H (Hielscher, Teltow, Germany) by three identical steps (amplitude 100%; 10 s) and incubated at room temperature for 1 h, followed by centrifugation (15,000× g at room temperature for 5 min) and the collection of the supernatant.
The purified PBMCs were washed with 1 mL of PBS. The cells were then incubated in the lysis mix and homogenized by ultrasonic treatment. Subsequently, the proteins were purified by precipitation using 600 µL precipitant (component of the 2-D Quant Kit, GE Healthcare, Uppsala, Sweden) per 250 µL of the sample, and a co-precipitant (component of the 2-D Quant Kit) in the same amount [
41]. The purified proteins were dissolved in 100 µL 9.8 M urea and solubilized for 1 h at room temperature. The protein concentration was determined using the Pierce BCA Protein Assay Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions, and using BSA as the standard. The samples were stored at −80 °C. The sample quality and the quality of the BCA-test results were confirmed by colloidal Coomassie-stained SDS gels. The staining procedure was performed according to Neuhoff [
63].
4.6. SDS-PAGE and Western Blot
The SDS-PAGE and Western blot analysis were performed as described [
32], with the following modifications. For the Western blot analysis, 20 µg protein or 40 µg protein per sample were applied. After the SDS-PAGE, the proteins were transferred to Immobilon-P
SQ membranes (Millipore GmbH, Schwalbach, Germany). The proteins were transferred by tank blot using the mini VE vertical electrophoresis system equipped with tank blot transfer units (GE Healthcare, Uppsala, Sweden). The bands were visualized using the Signal Fire ECL Reagent (Cell Signaling Technology, Danvers, MA, USA) and X-ray films (Agfa HealthCare, Mortsel, Belgium) in accordance with the manufacturer’s instructions. The X-ray films were digitized using a GS-700 imaging densitometer (Bio-Rad, Hercules, CA, USA). The densitometric analysis was performed using Quantity one software (Bio-Rad). Each reaction was performed in biological triplicates. The applied antibodies are listed in
Table 2.
The membranes were stripped using the following stripping buffer: 7.56 g Tris, 20 g SDS, 7.8 g 2-mercaptoethanol, adjustment to pH 9.5 using HCl, and H2O ad 1 l. Prior to its use, 0.1 g DTT was freshly added to 25 mL stripping buffer. The membranes were incubated at room temperature with gentle agitation for 45 min. After washing with tris-buffered saline with 0.1% Tween 20 (TBST), the membranes were incubated with blocking buffer for one hour, and another primary antibody was applied. The cleavage of the disulphide bridges by the reducing agents is very efficient at alkaline pH values, such that the removal of antibodies can be performed under mild conditions, allowing the multiple rehybridization of the membrane with antibodies.
In the case of the quantitative analysis of X-ray films, the values of different biological experiments were combined. First, the signals of the protein of interest were normalized to their associated alpha-tubulin values obtained from the same polyvinylidene difluoride (PVDF) membrane to level out the potential differences in the protein loading and transfer (intra-gel comparison). For the inter-gel comparison, the values were normalized by setting one value of one single experiment to 100 a. u. (usually the standard cell culture condition). After that, the values from the different experimental runs that were not set to 100 a. u. were compared. The antibodies used are specified in
Table 2.
For the correlation analysis between Cyr61 and keratin, we used Kendall’s Tau (kτ) and the software OriginPro (version 9.6.5.169, OriginLab Corporation, Northampton, USA).
4.7. Lentivirus-Mediated Knockdown of Cyr61 Gene Expression
The lentiviral pLKO.1 shRNA vectors targeted against human Cyr61 (
cyr61) were designed by The RNAi Consortium (Broad Institute of Harvard and Massachusetts Institute of Technology; GE Dharmacon, Uppsala, Sweden; Cat. No. RHS4533-EG3491 [
64]). The used vectors contained the following sequences:
Cyr61#1: TRCN0000118097 (Clone ID);
CCGGGCAAACAGAAATCAGGTGTTTCTCGAGAAACACCTGATTTCTGTTTGCTTTTTG (Forward Oligo Sequence);
AATTCAAAAAGCAAACAGAAATCAGGTGTTTCTCGAGAAACACCTGATTTCTGTTTGC (Reverse Oligo Sequence).
Cyr61#2: TRCN0000118098 (Clone ID);
CCGGCGCATCCTATACAACCCTTTACTCGAGTAAAGGGTTGTATAGGATGCGTTTTTG (Forward Oligo Sequence);
AATTCAAAAACGCATCCTATACAACCCTTTACTCGAGTAAAGGGTTGTATAGGATGCG (Reverse Oligo Sequence).
Cyr61#3: TRCN0000118099 (Clone ID);
CCGGCCGAACCAGTCAGGTTTACTTCTCGAGAAGTAAACCTGACTGGTTCGGTTTTTG (Forward Oligo Sequence);
AATTCAAAAACCGAACCAGTCAGGTTTACTTCTCGAGAAGTAAACCTGACTGGTTCGG (Reverse Oligo Sequence).
Cyr61#4: TRCN0000118100 (Clone ID);
CCGGCCCTTCTACAGGCTGTTCAATCTCGAGATTGAACAGCCTGTAGAAGGGTTTTTG (Forward Oligo Sequence);
AATTCAAAAACCCTTCTACAGGCTGTTCAATCTCGAGATTGAACAGCCTGTAGAAGGG (Reverse Oligo Sequence).
Cyr61#5: TRCN0000118101 (Clone ID);
CCGGCGAACCAGTCAGGTTTACTTACTCGAGTAAGTAAACCTGACTGGTTCGTTTTTG (Forward Oligo Sequence);
AATTCAAAAACGAACCAGTCAGGTTTACTTACTCGAGTAAGTAAACCTGACTGGTTCG (Reverse Oligo Sequence).
The pLKO.1 vectors harboring a scrambled non-target shRNA sequence (Addgene, Cambridge, MA, USA) served as a negative control (non-target control). The lentiviruses were made by transfection of HEK-293T packaging cells with these constructs by the use of a three-plasmid system, as described [
65]. The supernatants were harvested, sterile filtered, and used to infect MDA-MB-231 cells in the presence of 8 µg/mL polybrene (Sigma-Aldrich) as the final concentration, and with a multiplicity of infection (MOI) of 1. The pooled stable transfectants were established using an optimal final puromycin concentration of 0.5 µg/mL. These experiments were performed under the standard culture conditions for MDA-MB-231. After seven days, the knockdown efficiency was analysed by Western blot analysis and an enzyme-linked immunosorbent assay. Cyr61#4 and Cyr61#5 showed no detectable Cyr61 knockdown in MDA-MB-231 by Western blot analysis, and were not analysed further. The transfectants of Cyr61#2 and Cyr61#3 were used for the research of their biological functions with apoptosis and proliferation assays under standard cell culture conditions, and under hypoxia (1% O
2). The photomicrographs were taken using an Axiovert 25 inverted microscope (Carl Zeiss AG, Oberkochen, Germany).
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
The cell lysates and culture supernatants were clarified by centrifugation at 2500× g for 15 min. The recombinant human Cyr61 protein was purchased from Abnova (Taipei, Taiwan).
For the coating of the wells, the anti-Cyr61 antibody H2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was applied. The antibody was diluted 1:250 in DMEM with 10% FCS. The plate was incubated at 4 °C overnight, with gentle agitation. In order to remove the unbound antibody, the wells were washed with 100 µL volume three times. The first time, the wells were washed with PBS, followed by two steps using PBS with 0.02% Tween 20 (Roth, Karlsruhe, Germany). Next, the unspecific binding was blocked with blocking buffer (5% nonfat dry milk [Roth], in PBS with 0.02% Tween) using 100 µL blocking buffer per well. Next, three washing steps using 100 µL volume each, as described, were performed. For the incubation with cell culture supernatant, 2.5 µL of the sample was diluted in 97.5 µL DMEM with 10% FCS. This was followed by three washing steps, as described. Next, the anti-Cyr61 antibody H78 (Santa Cruz Biotechnology) was added to the wells. The anti-Cyr61 antibody was diluted 1:500 in DMEM with 10% FCS. This was followed by three washing steps. For the detection of Cyr61, a polyclonal goat anti-rabbit immunoglobulin antibody coupled with horseradish peroxidase (DAKO, Glostrup, Denmark) was diluted 1:250 with blocking buffer. The reaction was incubated at room temperature for one hour, with gentle agitation. This was followed by three washing steps. Next, 100 µL 3,3′,5,5′-tetramethylbenzidine (TMB) one-component horseradish peroxidase (HRP) microwell substrate (Bethyl Laboratories, Montgomery, PA, USA) was added to each well. The incubation was carried out whilst protected from light at room temperature for 15 min. The reaction was stopped by the addition of 100 µL Stop Solution for TMB Substrates (Immunochemistry Technologies, Bloomington, IN, USA) and incubation in the dark with gentle agitation for 15 min. The extinction at 450/620 nm was detected using the ELISA reader NanoQuant infinite M200 pro (Tecan, Männedorf, Switzerland). The OD values were converted to Cyr61 concentrations using recombinant and purified Cyr61 protein as a standard.
4.9. Proliferation and Apoptosis Assays
For the analysis of the cell proliferation and apoptosis, MDA-MB-231 Cyr61 knock down cells (Cyr61#2 and Cyr61#3) and the corresponding non-target control cells were applied. The cell proliferation was analysed by the incorporation of 5-bromo-2′-deoxyuridine (BrdU) into the DNA of the proliferating cells using the BrdU Cell Proliferation Assay Kit (Cell Signaling Technology). The cells were plated in a concentration of 30,000 cells per well in 96-well TC plates, Standard, F (Sarstedt, Nümbrecht, Germany) and cultured either under the standard cell culture conditions or under 1% O2 for 72 h. Subsequently, the BrdU solution was added to the cells and incubated on the cells for 24 h. Next, 100 µL of the Fixing/Denaturing Solution was added to each well and incubated for 30 min, followed by the incubation of 100 µL of the detection antibody solution per well for 1 h. After the washing of the wells, the HRP-conjugated secondary antibody solution was applied to the wells and incubated at room temperature for 30 min. For the detection, 100 µL 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was applied to each well and incubated at room temperature whilst protected from light. After the addition of the stop solution, the absorbance was detected at 450 nm using the ELISA reader NanoQuant infinite M200 pro.
The apoptosis of the cells was analysed by annexin V staining using APC Annexin V (BioLegend, San Diego, CA, USA). The cells were cultured under the standard culture conditions, or under 1% O2. After that, the cells were washed twice with Cell Staining Buffer (BioLegend) and resuspended in Annexin V Binding Buffer (BioLegend) to a final concentration of 1 × 106 cells/mL. The cell suspension was supplemented with 5 µL APC Annexin V per 100 µL of the cell suspension. After incubation for 15 min at room temperature, protected from light, 400 µL Annexin V Binding Buffer was added to each sample and analysed by flow cytometry (FACSCanto II, Franklin Lakes, NJ, USA).
4.10. Size Based CTC Enrichment
We applied a marker-independent separation device (Parsortix, ANGLE PLC, Surrey, UK) for the tumor cell enrichment. The Parsortix system uses a micro-fluidic technology in the form of a disposable cassette (Cell separation cassette cc3R, Parsortix) to capture CTCs out of the blood from cancer patients. In total, 4 ml blood was collected into BD Vacutainers (BD Belliver Industrial Estate, Plymouth, UK). The blood was pumped automatically through the cassette. The cassette enriches CTCs based on their larger size (≥10 µm) compared to other blood components. In order to reduce the background noise, the cassette was automatically washed with PBS (Life Technologies). The isolated tumor cells were harvested, cytospun on a slide (SuperFrost/Plus, Glaswarenfabrik Karl Hecht KG, Sondheim, Germany), and stained, as described below. For the cell line spiking experiments with blood from healthy persons, fresh blood was received from the Institute for Transfusion Medicine, University Medical Center Hamburg-Eppendorf. The clinical samples from breast cancer patients were drawn from the Department of Gynecology.
4.11. Purification of the Bone Marrow Specimens and Peripheral Blood Samples
Bone marrow was aspirated bilaterally from both the anterior and posterior iliac crests (10 mL/site) from breast cancer patients or healthy volunteers. The following procedures were accomplished under sterile conditions. The bone marrow aspirates were washed in Hanks’ Balanced Salt solution (HBSS) (Biochrom AG, Berlin, Germany), diluted in Dulbecco’s phosphate-buffered saline (DPBS) (Life Technologies), and separated by density centrifugation using Ficoll Paque Plus (GE Healthcare, Munich, Germany). Mononuclear cells were collected from the interphase layer and washed twice in phosphate buffered saline (PBS) with 10% fetal calf serum (Biological Industries, Kibbutz Beit Haemek, Israel). The cytospins were prepared by centrifuging the bone marrow mononuclear cells onto glass slides (Superfrost plus, Glaswarenfabrik Karl Hecht KG, Sondheim, Germany; 7 × 105 mononuclear cells per slide). The slides were air-dried overnight and stored at −80 °C.
For the experiments using peripheral blood from healthy individuals, the PBMC were separated by Ficoll density centrifugation. The obtained PBMC were collected from the interphase layer, washed with PBS, and either applied for the spiking experiments or analyzed by Western blot analysis. For the spiking experiments, the cell lines were spiked into the blood or bone marrow from healthy individuals and processed as described.
4.12. Immunocytochemical Cyr61 Detection in Cell Lines, Blood and Bone Marrow Samples
The cell lines were spiked into the blood or bone marrow samples of healthy control persons. The immunocytochemical double staining was performed by the application of the anti-Cyr61 antibody (H78) in combination with a Cytokeratin-specific antibody cocktail. The Cytokeratin antibody cocktail consisted of anti-pan-keratin antibodies (mouse monoclonal, clones AE1/AE3; Merck) and anti-pan-keratin antibody C11 (mouse monoclonal; Cell Signaling Technology Cat# 4545). The direct conjugates of AE1/AE3-Alexa Fluor488 (Thermo Fisher Scientific Cat# 53-9003-82) and C11-Alexa Fluor488 (Cell Signaling Technology Cat# 4523) were used when stated for the individual experiments. The detection of normal blood cells was performed using the anti-CD45 antibody coupled with Alexa Fluor 647 (BioLegend, Cat# 304018), when stated.
The slides were thawed 30 min prior to their incubation. The cells were permeabilized for 10 min with 1% Triton X-100 in PBS. A washing step was followed by the blocking of unspecific binding using AB-Serum (Biotest, Dreieich, Germany, Cat. No. 805 135) (10% in PBS) for 20 min. The primary antibody against Cyr61 was added in a 1:50 dilution and incubated at room temperature for 1 h. The slides were washed again three times with PBS, and Alexa 546 or Alexa 532 goat anti-rabbit secondary antibody (Molecular Probes, Eugene, OR, USA; Thermo Fisher Scientific Cat# A-11035, Thermo Fisher Scientific Cat# A-11009) was applied. After three washing steps with PBS, the Cytokeratin specific antibodies were applied and incubated for 60 min. For the AE1/AE3 C11 antibody cocktail, the dilution was 1:700 for the AE1/AE3, and the C11 was diluted 1:200. The residual Cytokeratin-specific antibodies were removed by three washing steps with PBS. If unconjugated Cytokeratin specific antibodies were applied, the secondary Alexa 488 rabbit anti-mouse fluorochrome antibody (Molecular Probes; Molecular Probes Cat# A-11059) was added in a 1:200 dilution in 10% AB-Serum and incubated for 45 min. After another washing step (3 × with PBS), the slides were covered with Vectashield Mounting Medium containing Dapi (Vector Laboratories, Burlingame, USA). The staining controls were run in parallel, using dilution media instead of the primary and secondary antibody. The slides were evaluated manually using the microscope Axioplan 2 (Carl Zeiss AG, Oberkochen, Germany).
4.13. Cyr61 Immunohistochemical Staining
For the immunohistochemical (IHC) staining, the anti-Cyr61 antibody (H78) rabbit polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was applied. This antibody was applied previously by another working group for TMA staining in prostate cancer [
52]. Paraffin-embedded specimens on microscope slides of breast cancer patients were applied. The paraffin wax was removed by the incubation of the specimen at 60 °C for two hours, followed by the incubation of the samples twice in xylene for 10 min each. In order to remove the xylene, the slides were incubated in 99% ethanol, followed by incubation in 96% ethanol and in 80% ethanol. After a brief washing step in water, the samples were autoclaved at 120 °C in citrate buffer (pH 6.0) for 5 min. Thereafter, the sections were rinsed with TBST for 5 min. A peroxidase treatment was performed using the Dako REAL Peroxidase-Blocking Solution (DAKO, Cat. No. S202386-2) for five minutes. After a brief washing step with TBST, the anti-Cyr61 antibody was applied. The antibody was used in a 1:750 dilution using the Dako Antibody Diluent (DAKO) and incubated at 4 °C overnight. Subsequently, three 3-min washing steps with TBST were performed. For the detection of the primary antibody, labelled polymer-HRP and the secondary antibody was used from the DAKO REAL Detection system Peroxidase/DAB (DAKO #K5001) according to the manufacturer’s instructions. For the chromogenic detection, 3,3’-diaminobenzidine (DAB) was applied. After a brief washing step, the nuclei were visualized by haematoxylin staining (Merck, Darmstadt, Germany). For the preservation of the specimen, Eukitt mounting medium (Kindler, Freiburg, Germany) was used.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}