
Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
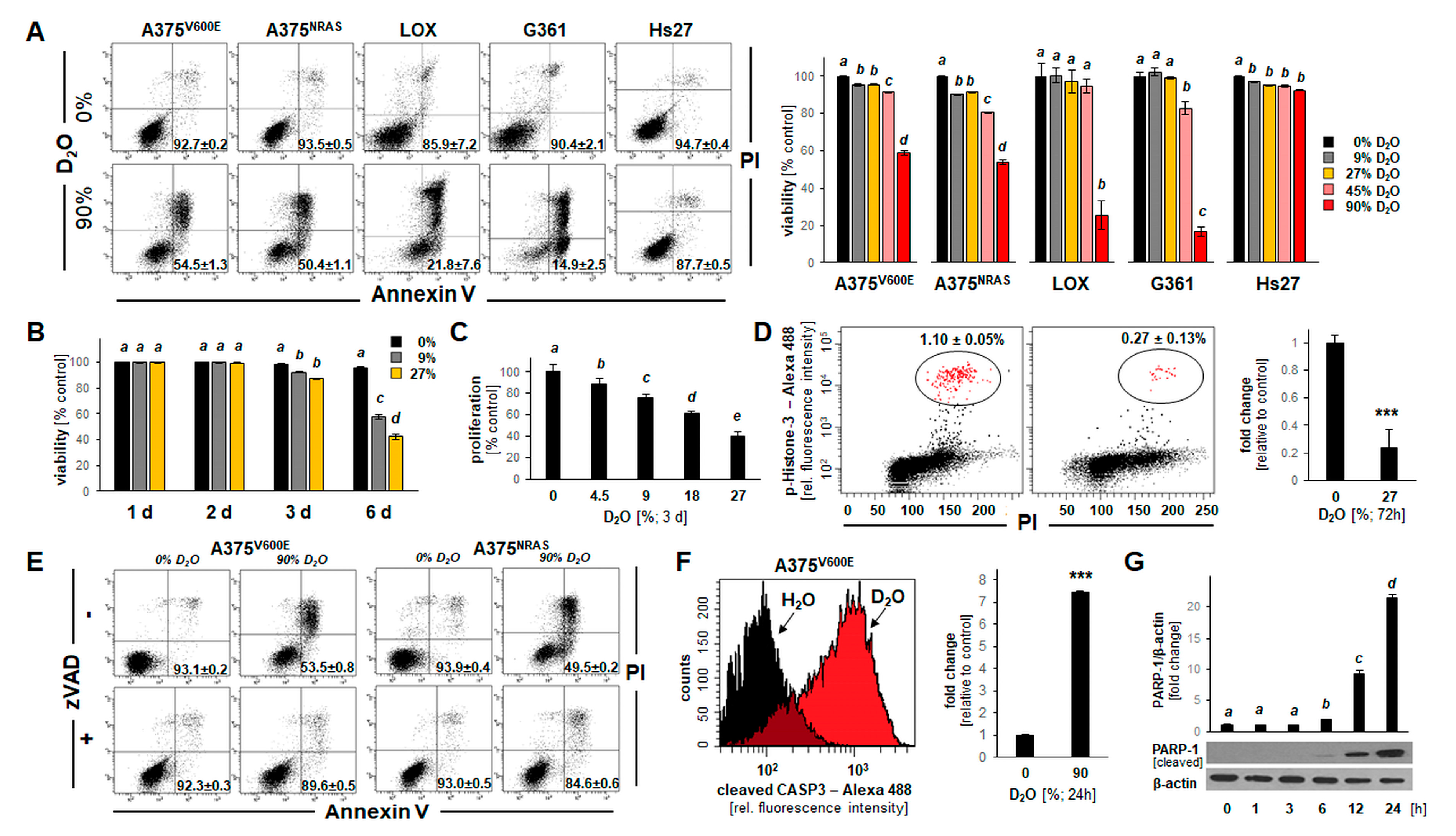
2.1. D2O Exposure Induces Apoptotic Cell Death in a Panel of Cultured Human Malignant Melanoma Cells
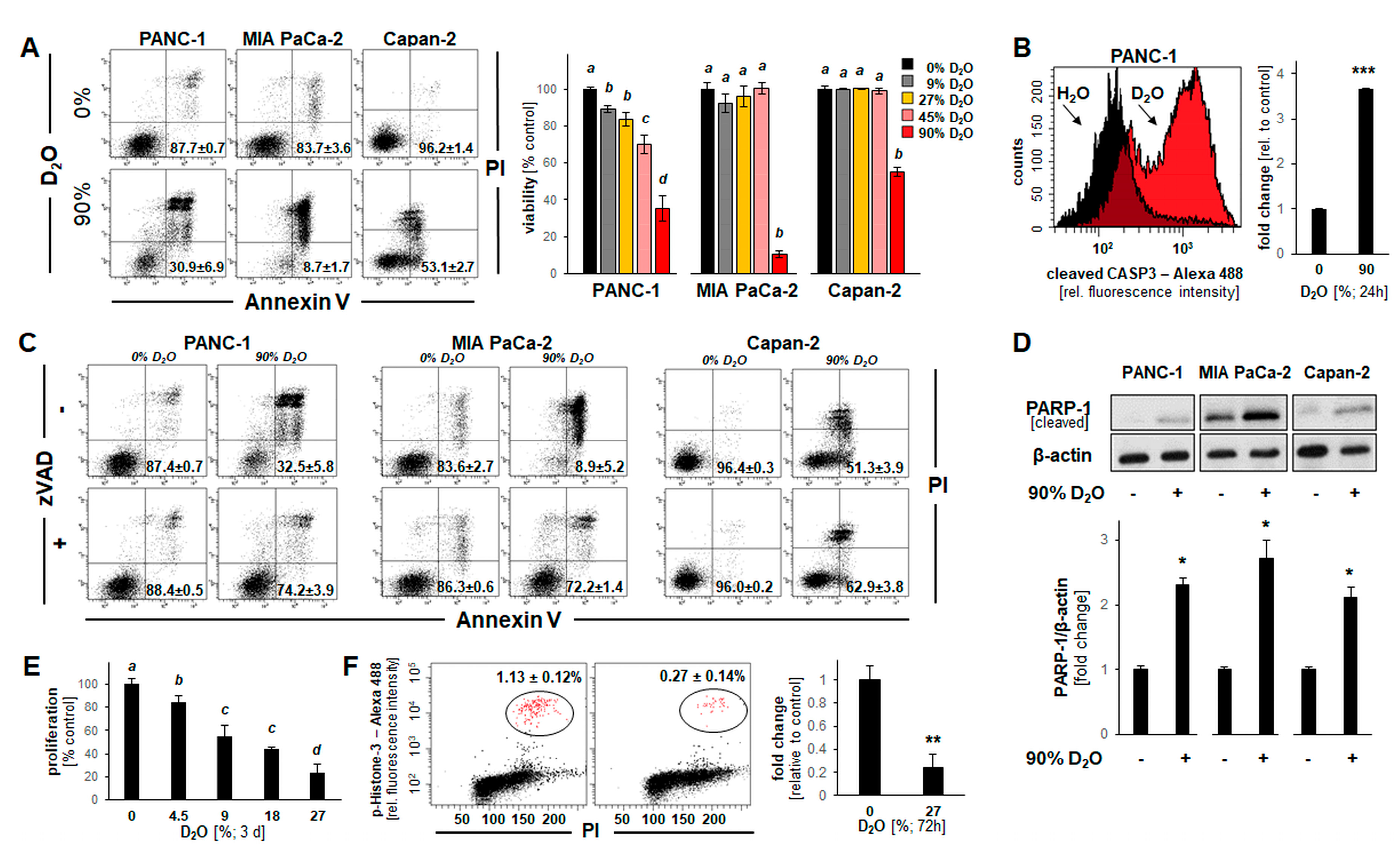
2.2. D2O Exposure Induces Apoptotic Cell Death in a Panel of Human Pancreatic Ductal Adenocarcinoma Cells (PANC-1, MIA PaCa-2, and Capan-2)
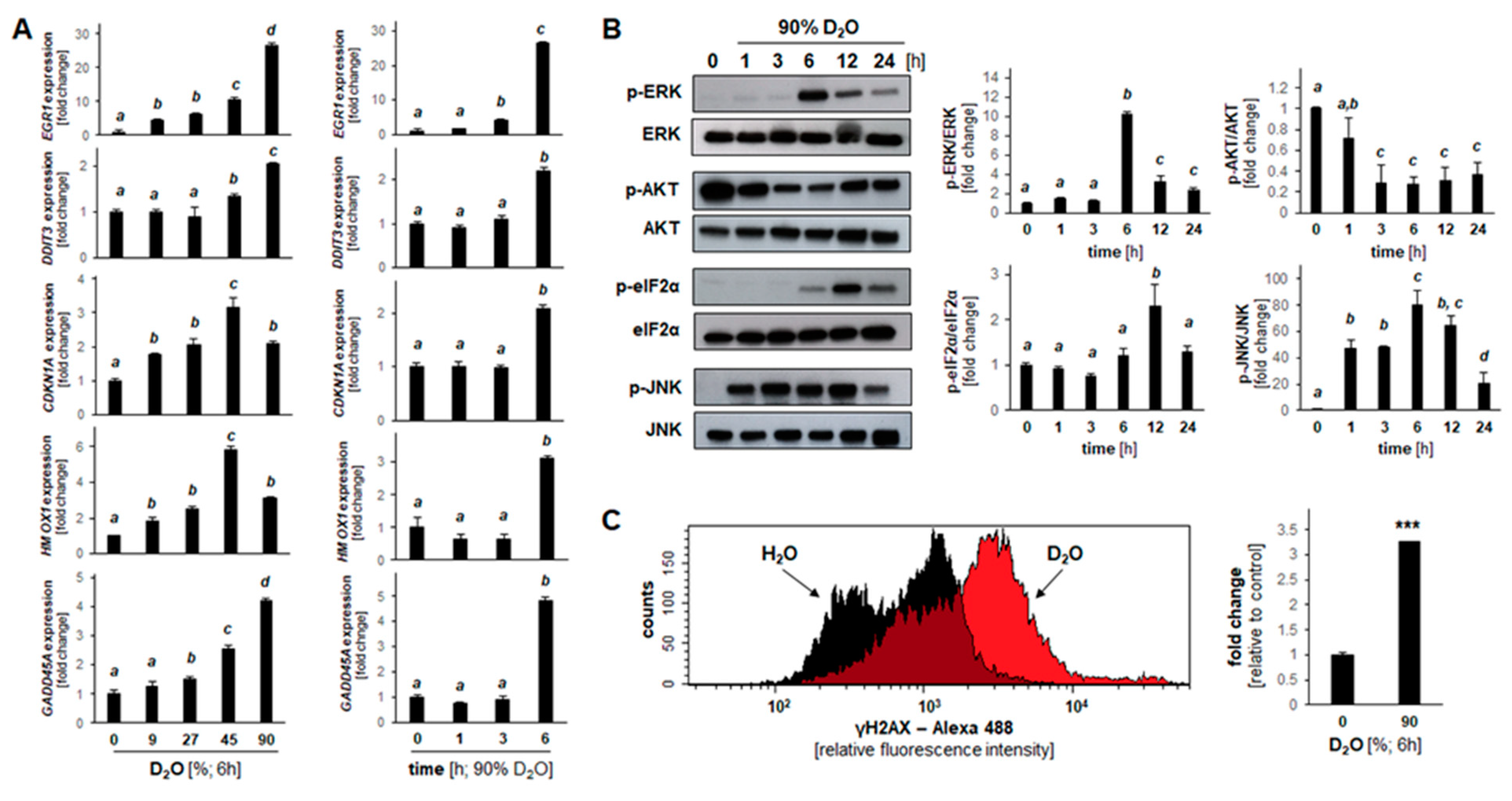
2.3. Array Analysis Reveals D2O-Induced Early Stress Response Gene Expression Changes Observable in Both Cultured Malignant Melanoma (A375) and Pancreatic Ductal Adenocarcinoma (PANC-1) Cells
2.4. D2O Modulates Stress Response Gene Expression and Protein Phosphorylation in A375 Melanoma Cells
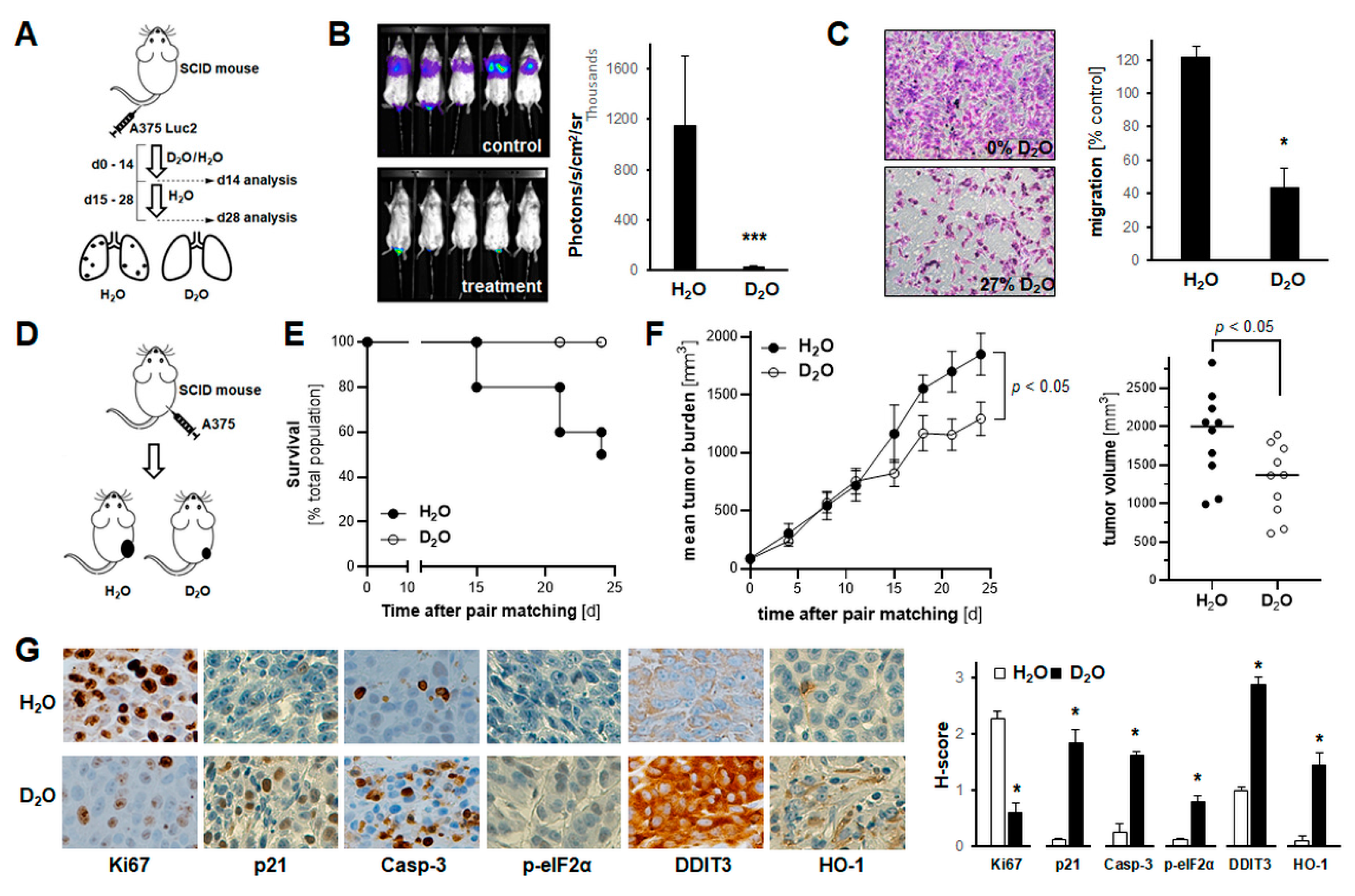
2.5. Systemic Administration of D2O Impairs Metastasis and Tumor Growth in SCID Mouse Models of Human Malignant Melanoma
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. RNA Extraction and Single Reverse Transcrition Quantitative Polymerase Chain Reaction (RT-qPCR)
4.4. Immunoblot Detection
4.5. Flow Cytometric Analysis of Cell Viability
4.6. Cell Proliferation Assay
4.7. M-Phase Quantification by Phospho-Histone H3 (Ser10) Flow Cytometry
4.8. Caspase-3 Activation Assay
4.9. Cytometric Assessment of Histone H2AX Phosphorylation
4.10. Comparative RT2 ProfilerTM PCR Gene Expression Array Analysis
4.11. Transwell Invasion Assay
4.12. Metastasis Model in SCID Mice
4.13. Xenograft Model in SCID Mice
4.14. Immunohistochemistry
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urey, H.C.; Brickwedde, F.G.; Murphy, G.M. A hydrogen isotope of mass 2. Phys. Rev. 1932, 39, 164–165. [Google Scholar] [CrossRef]
- Barbour, H.G.; Allen, E. Tumor Growth in Mice One-Fifth Saturated with Deuterium Oxide (Heavy Water). Cancer Res. 1938, 32, 440–446. [Google Scholar]
- Laissue, J.A.; Burki, H.; Berchtold, W. Survival of tumor-bearing mice exposed to heavy water or heavy water plus methotrexate. Cancer Res. 1982, 42, 1125–1129. [Google Scholar]
- Laissue, J.A.; Altermatt, H.J.; Bally, E.; Gebbers, J.O. Protection of mice from whole body gamma irradiation by deuteration of drinking water: Hematologic findings. Exp. Hematol. 1987, 15, 177–180. [Google Scholar] [PubMed]
- Altermatt, H.J.; Gebbers, J.O.; Laissue, J.A. Heavy-Water Delays Growth of Human Carcinoma in Nude-Mice. Cancer 1988, 62, 462–466. [Google Scholar] [PubMed]
- Altermatt, H.J.; Gebbers, J.O.; Laissue, J.A. Heavy water enhances the antineoplastic effect of 5-fluoro-uracil and bleomycin in nude mice bearing human carcinoma. Int. J. Cancer 1990, 45, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Gaeng, D.P.; Geiser, M.; Cruz-Orive, L.M.; Larsen, S.E.; Schaffner, T.; Laissue, J.A.; Altermatt, H.J. Paradoxical effects of bleomycin and heavy water (D2O) in mice. Int. J. Cancer 1995, 62, 784–790. [Google Scholar] [CrossRef]
- Wallace, S.A.; Mathur, J.N.; Allen, B.J. The Influence of Heavy-Water on Boron Requirements for Neutron-Capture Therapy. Med. Phys. 1995, 22, 585–590. [Google Scholar] [CrossRef]
- Takeda, H.; Nio, Y.; Omori, H.; Uegaki, K.; Hirahara, N.; Sasaki, S.; Tamura, K.; Ohtani, H. Mechanisms of cytotoxic effects of heavy water (deuterium oxide: D2O) on cancer cells. Anti Cancer Drugs 1998, 9, 715–725. [Google Scholar] [CrossRef]
- Kushner, D.J.; Baker, A.; Dunstall, T.G. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can. J. Physiol. Pharmacol. 1999, 77, 79–88. [Google Scholar] [CrossRef]
- Schloerb, P.R.; Friis-Hansen, B.J.; Edelman, I.S.; Solomon, A.K.; Moore, F.D. The measurement of total body water in the human subject by deuterium oxide dilution; with a consideration of the dynamics of deuterium distribution. J. Clin. Invest. 1950, 29, 1296–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagojevic, N.; Storr, G.J.; Alien, B.; Hatanaka, H.; Nakagawa, Y. Role of heavy water in boron neutron capture therapy. In Topics in Dosimetry & Treatment Planning for Boron Neutron Capture Therapy; Advanced Medical Publishing: Madison, WI, USA, 1994; pp. 125–134. [Google Scholar]
- Maybury, R.H.; Katz, J.J. Protein Denaturation in Heavy Water. Nature 1956, 177, 629–630. [Google Scholar] [CrossRef]
- Kresheck, G.C.; Schneider, H.; Scheraga, H.A. The effect of D2-O on the thermal stability of proteins. Thermodynamic parameters for the transfer of model compounds from H2-O to D2-O. J. Phys. Chem. 1965, 69, 3132–3144. [Google Scholar] [CrossRef]
- Baghurst, P.A.; Sawyer, W.H.; Nichol, L.W. Effect of D2O on Association of Beta-Lactoglobulin A. J. Biol. Chem. 1972, 247, 3199–3204. [Google Scholar] [CrossRef]
- Zimmermann, A.; Keller, H.U.; Cottier, H. Heavy water (D2O)-induced shape changes, movements and F-actin redistribution in human neutrophil granulocytes. Eur. J. Cell. Biol. 1988, 47, 320–326. [Google Scholar] [PubMed]
- Cioni, P.; Strambini, G.B. Effect of heavy water on protein flexibility. Biophys. J. 2002, 82, 3246–3253. [Google Scholar] [CrossRef] [Green Version]
- Itoh, T.J.; Sato, H. The effects of deuterium oxide (2H2O) on the polymerization of tubulin in vitro. Biochim. Biophys. Acta 1984, 800, 21–27. [Google Scholar] [CrossRef]
- Prodhom, B.; Pietrobon, D.; Hess, P. Direct Measurement of Proton-Transfer Rates to a Group Controlling the Dihydropyridine-Sensitive Ca-2+ Channel. Nature 1987, 329, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, S.; Sampson, C.A.; Liepins, A. Effects of deuterium oxide (D2O) on the development of hypertension and Ca2+ homeostasis in spontaneously hypertensive rats. J. Hypertens. 1990, 8, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, J.; Schroeter, D.; Paweletz, N. Derangement of microtubule arrays in interphase and mitotic PtK2 cells treated with deuterium oxide (heavy water). J. Cell. Sci. 1991, 98, 463–473. [Google Scholar]
- Chakrabarti, G.; Kim, S.; Gupta, M.L.; Barton, J.S.; Himes, R.H. Stabilization of tubulin by deuterium oxide. Biochemistry 1999, 38, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Chaen, S.; Yamamoto, N.; Shirakawa, B.; Sugi, H. Effect of deuterium oxide on actomyosin motility in vitro. Biochim. Biophys. Acta Bioenerg. 2001, 1506, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Hirakura, Y.; Sugiyama, T.; Takeda, M.; Ikeda, M.; Yoshioka, T. Deuteration as a tool in investigating the role of protons in cell signaling. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 218–225. [Google Scholar] [CrossRef]
- Radisavljevic, Z. AKT as Locus of Hydrogen Bond Network in Cancer. J. Cell. Biochem. 2018, 119, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Park, H.J.; Cho, N.; Kim, H.P. Aquaporin 11-Dependent Inhibition of Proliferation by Deuterium Oxide in Activated Hepatic Stellate Cells. Molecules 2018, 23, 3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manson, L.A.; Defendi, V.; Hartzell, R.W.; Kritchevsky, D. Effect of deuterium oxide on growth of HeLa, L and L-5178Y cells. Proc. Soc. Exp. Biol. Med. 1960, 105, 481–483. [Google Scholar] [CrossRef]
- Schroeter, D.; Lamprecht, J.; Eckhardt, R.; Futterman, G.; Paweletz, N. Deuterium-Oxide (Heavy-Water) Arrests the Cell-Cycle of Ptk2 Cells during Interphase. Eur. J. Cell Biol. 1992, 58, 365–370. [Google Scholar]
- Hartmann, J.; Bader, Y.; Horvath, Z.; Saiko, P.; Grusch, M.; Illmer, C.; Madlener, S.; Fritzer-Szekeres, M.; Heller, N.; Alken, R.G.; et al. Effects of heavy water (D2O) on human pancreatic tumor cells. Anticancer Res. 2005, 25, 3407–3411. [Google Scholar]
- Bahk, J.Y.; Lee, J.H.; Chung, H.S.; Lee, H.Y.; Chung, B.C.; Park, M.S.; Min, S.K.; Kim, M.O. Anticancer effect of deuterium oxide on a bladder cancer cell related to bcl-2 and bax. J. Ind. Eng. Chem. 2007, 13, 501–507. [Google Scholar]
- Kumar, N.; Attri, P.; Yadav, D.K.; Choi, J.; Choi, E.H.; Uhm, H.S. Induced apoptosis in melanocytes cancer cell and oxidation in biomolecules through deuterium oxide generated from atmospheric pressure non-thermal plasma jet. Sci. Rep. 2014, 4, 7589. [Google Scholar] [CrossRef] [Green Version]
- Kleemann, J.; Reichenbach, G.; Zoller, N.; Jager, M.; Kaufmann, R.; Meissner, M.; Kippenberger, S. Heavy Water Affects Vital Parameters of Human Melanoma Cells in vitro. Cancer Manag. Res. 2020, 12, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Wondrak, G.T. (Ed.) Stress Response Pathways in Cancer: From Molecular Targets to Novel Therapeutics, 1st ed.; Springer: Heidelberg, Germany, 2015; pp. 1–446. [Google Scholar]
- Ueda, Y.; Richmond, A. NF-kappaB activation in melanoma. Pigment. Cell Res. 2006, 19, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Boone, D.N.; Qi, Y.; Li, Z.; Hann, S.R. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.L.; Qiao, S.; Lesson, J.L.; Rojo de la Vega, M.; Park, S.L.; Seanez, C.M.; Gokhale, V.; Cabello, C.M.; Wondrak, G.T. The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J. Biol. Chem. 2015, 290, 1623–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, M.; Stojanovic, N.; Christian, J.; Paul, M.C.; Stauber, R.H.; Schmid, R.M.; Hacker, G.; Kramer, O.H.; Saur, D.; Schneider, G. MYC and EGR1 synergize to trigger tumor cell death by controlling NOXA and BIM transcription upon treatment with the proteasome inhibitor bortezomib. Nucleic Acids Res. 2014, 42, 10433–10447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jiang, G.; Mao, P.; Zhang, J.; Zhang, L.; Liu, L.; Wang, J.; Owusu, L.; Ren, B.; Tang, Y.; et al. Down-regulation of GADD45A enhances chemosensitivity in melanoma. Sci. Rep. 2018, 8, 4111. [Google Scholar] [CrossRef] [PubMed]
- Bader, Y.; Hartmann, J.; Horvath, Z.; Saiko, P.; Grusch, M.; Madlener, S.; Maier, S.; Oehler, L.; Fritzer-Szekeres, M.; Heller, N.; et al. Synergistic effects of deuterium oxide and gemcitabine in human pancreatic cancer cell lines. Cancer Lett. 2008, 259, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.N.; Huang, T.F.; Hung, C.F.; Chou, K.H.; Tsai, Y.J.; Wu, W.B. Activation of c-Jun N-teraminal kinase is essential for mitochondrial membrane potential change and apoptosis induced by doxycycline in melanoma cells. Br. J. Pharm. 2010, 160, 1171–1184. [Google Scholar] [CrossRef] [Green Version]
- Kwong, L.N.; Davies, M.A. Navigating the therapeutic complexity of PI3K pathway inhibition in melanoma. Clin. Cancer Res. 2013, 19, 5310–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikita, H.; Saito, K. Proton transfer reactions and hydrogen-bond networks in protein environments. J. R. Soc. Interface 2014, 11, 20130518. [Google Scholar] [CrossRef] [PubMed]
- Edington, B.V.; Whelan, S.A.; Hightower, L.E. Inhibition of heat shock (stress) protein induction by deuterium oxide and glycerol: Additional support for the abnormal protein hypothesis of induction. J. Cell Physiol. 1989, 139, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, J.; Schroeter, D.; Paweletz, N. Mitosis arrested by deuterium oxide. Light microscopic, immunofluorescence and ultrastructural characterization. Eur. J. Cell. Biol. 1990, 51, 303–312. [Google Scholar]
- Bauer, A.L.; Jachimczak, P.; Blesch, A.; Baur, J.; Hessdorfer, B.; Haase, A.; Bogdahn, U. Selective Killing and Growth Arrest of Malignant-Tumor Cells by Deuterium-Oxide (D2O). Tumordiagnost. Ther. 1995, 16, 61–68. [Google Scholar]
- Zlatska, A.; Zlatskyi, I.; Syroeshkin, A. The role of deuterium in the biological properties of human adipose derived mesenchymal stem cells in vitro. FEBS Open Biol. 2018, 8, 148. [Google Scholar]
- Cabello, C.M.; Lamore, S.D.; Bair, W.B.; Qiao, S.; Azimian, S.; Lesson, J.L.; Wondrak, G.T. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest. New Drugs 2012, 30, 1289–1301. [Google Scholar] [CrossRef]
- Jandova, J.; Wondrak, G.T. Genomic GLO1 deletion modulates TXNIP expression, glucose metabolism, and redox homeostasis while accelerating human A375 malignant melanoma tumor growth. Redox Biol. 2020, 39, 101838. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B.; Ley, S.; Lamore, S.D.; Azimian, S.; Wondrak, G.T. The experimental chemotherapeutic N6-furfuryladenosine (kinetin-riboside) induces rapid ATP depletion, genotoxic stress, and CDKN1A(p21) upregulation in human cancer cell lines. Biochem. Pharmacol. 2009, 77, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Perer, J.; Jandova, J.; Fimbres, J.; Jennings, E.Q.; Galligan, J.J.; Hua, A.; Wondrak, G.T. The sunless tanning agent dihydroxyacetone induces stress response gene expression and signaling in cultured human keratinocytes and reconstructed epidermis. Redox Biol. 2020, 36, 101594. [Google Scholar] [CrossRef]
- Jandova, J.; Perer, J.; Hua, A.; Snell, J.A.; Wondrak, G.T. Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo. Cancers 2020, 12, 1369. [Google Scholar] [CrossRef]
- Park, S.L.; Justiniano, R.; Williams, J.D.; Cabello, C.M.; Qiao, S.; Wondrak, G.T. The Tryptophan-Derived Endogenous Aryl Hydrocarbon Receptor Ligand 6-Formylindolo[3,2-b]Carbazole Is a Nanomolar UVA Photosensitizer in Epidermal Keratinocytes. J. Invest. Dermatol. 2015, 135, 1649–1658. [Google Scholar] [CrossRef] [Green Version]
- Cabello, C.M.; Bair, W.B.; Lamore, S.D.; Ley, S.; Bause, A.S.; Azimian, S.; Wondrak, G.T. The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth. Free Radic. Biol. Med. 2009, 46, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Bair, W.B.; Cabello, C.M.; Uchida, K.; Bause, A.S.; Wondrak, G.T. GLO1 overexpression in human malignant melanoma. Melanoma Res. 2009, 20, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Long, M.; Tao, S.; Rojo de la Vega, M.; Jiang, T.; Wen, Q.; Park, S.L.; Zhang, D.D.; Wondrak, G.T. Nrf2-dependent suppression of azoxymethane/dextran sulfate sodium-induced colon carcinogenesis by the cinnamon-derived dietary factor cinnamaldehyde. Cancer Prev. Res. 2015, 8, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandova, J.; Hua, A.B.; Fimbres, J.; Wondrak, G.T. Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers 2021, 13, 605. https://doi.org/10.3390/cancers13040605
Jandova J, Hua AB, Fimbres J, Wondrak GT. Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers. 2021; 13(4):605. https://doi.org/10.3390/cancers13040605
Chicago/Turabian StyleJandova, Jana, Anh B. Hua, Jocelyn Fimbres, and Georg T. Wondrak. 2021. "Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma" Cancers 13, no. 4: 605. https://doi.org/10.3390/cancers13040605