
Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression


Abstract
:Simple Summary
Abstract
1. Introduction
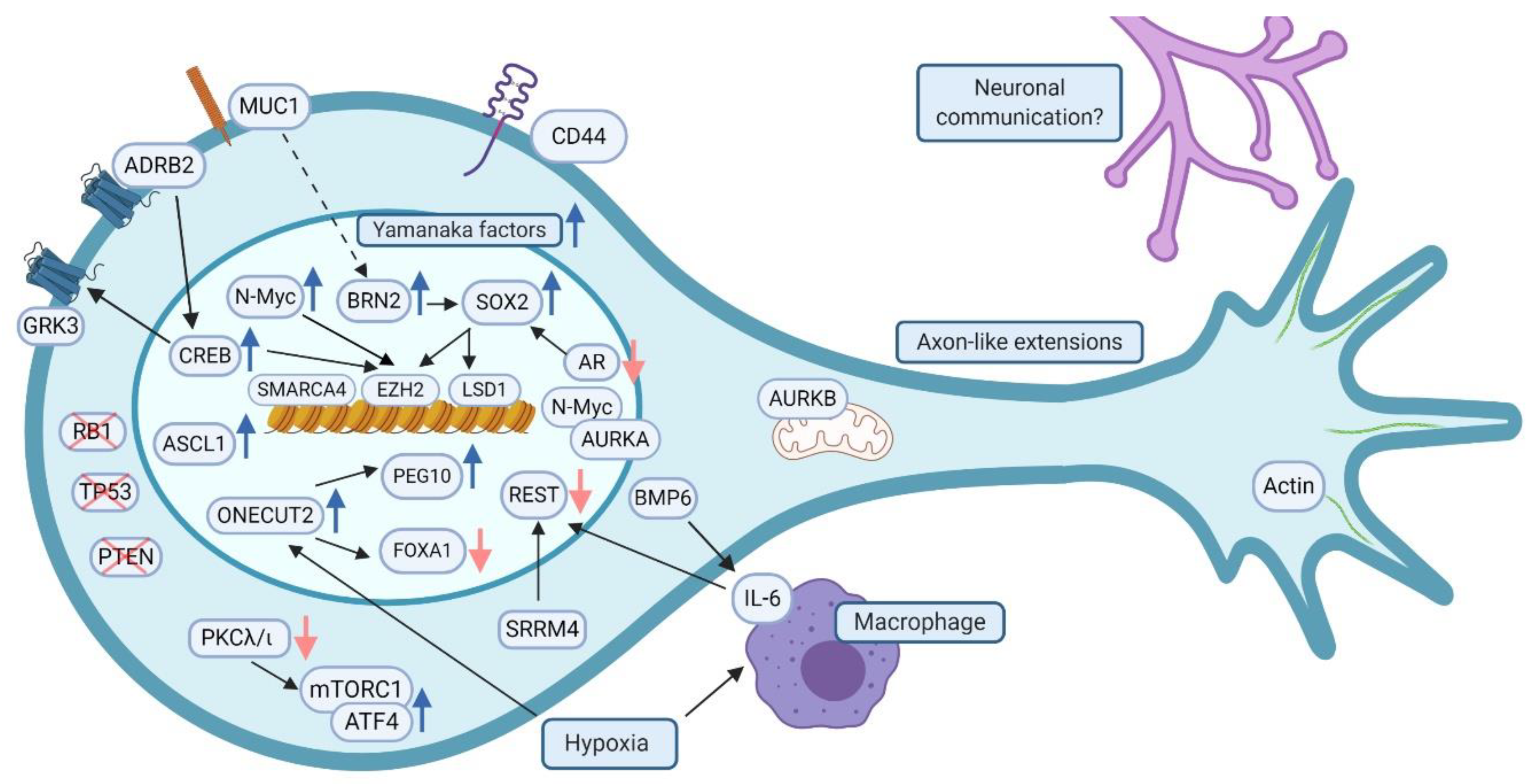
2. Pathways and Proteins Regulating t-NEPC Phenotype and Their Functional Relation to Neurodevelopmental Processes Neurogenesis, Axonogenesis and Synaptogenesis
2.1. Lack of Tumor Suppressors TP53, RB1 and PTEN
2.2. Transcription Factors Driving Treatment Resistance and NEPC Phenotype
2.2.1. BRN2 and SOX2
2.2.2. ASCL1
2.2.3. ONECUT2
2.2.4. REST
2.2.5. FOXM1
2.2.6. N-Myc
2.3. Mitotic Spindle Proteins Aurora Kinases A and B
2.4. Epigenetic Modulators and Chromatin Remodelling Complex Members and Their Regulators
2.4.1. EZH2, CREB and GRK3
2.4.2. SMARCA4
2.5. Receptors
2.5.1. Steroid Receptors
2.5.2. Adrenergic Receptors
2.5.3. ROR2
2.6. RNA Splicing Factors; the RNA Splicing Factor Serine/Arginine Repetitive Matrix 4, SRRM4
2.7. PKCλ/ι and Serine Synthesis Modulators
2.8. Cell Surface Membrane-Anchored Proteins
2.8.1. MUC1
2.8.2. CD44
3. Pluripotency Transcription Factors and Neuroendocrine Plasticity
4. The Role of Cell-Cell Communication Networks and Tumor Microenvironment in t-NEPC
4.1. Formation of Tunneling Microtubes
4.2. Microenvironmental Factors, Prostate-Neuron Interactions in the Tumor Microenvironment and the Hypothesis of Prostate—Neuron Cell Fusions
5. Emerging Targets and Current Clinical Trials under Investigation for NEPC
6. Future Directions
6.1. Are Neurodevelopmental Processes Truly Activated in Neuroendocrine Differentiation?
6.2. What Is the Role of Tumor Microenvironment in Neuron-Like Phenotypic Plasticity, Do Neuroendocrine Prostate Cancer Cells Connect to Neurons and Are They Able to Establish Functional Cancer-Neuron Connections?
6.3. Does the Neuroendocrine Phenotypic Plasticity in Prostate Cancer Harbor Cellular Transdiffention Processes Similar as Are Seen in the Induced Pluripotent Stem Cells and Their Transdifferentiation to Functional Neurons?
6.4. What Are the Key Players for the Phenotypic Switch? Are There Any Modulators of the Transdifferenation Process Available beyond Transcription Factors?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beltran, H.; Tagawa, S.T.; Park, K.; Macdonald, T.; Milowsky, M.I.; Mosquera, J.M.; Rubin, M.A.; Nanus, D.M. Challenges in Recognizing Treatment-Related Neuroendocrine Prostate Cancer. J. Clin. Oncol. 2012, 30, e386–e389. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.; Logothetis, C.J.; Maity, S.N. Understanding the Lethal Variant of Prostate Cancer: Power of Examining Extremes: Figure. Cancer Discov. 2011, 1, 466–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 1–10. [Google Scholar] [CrossRef]
- Chedgy, E.C.; Vandekerkhove, G.; Herberts, C.; Annala, M.; Donoghue, A.J.; Sigouros, M.; Ritch, E.; Struss, W.; Konomura, S.; Liew, J.; et al. Biallelic tumour suppressor loss and DNA repair defects in de novo small-cell prostate carcinoma. J. Pathol. 2018, 246, 244–253. [Google Scholar] [CrossRef]
- Suzuki, K.; Terakawa, T.; Jimbo, N.; Inaba, R.; Nakano, Y.; Fujisawa, M. Clinical Features of Treatment-Related Neuro-endocrine Prostate Cancer: A Case Series. Anticancer Res. 2020, 40, 3519–3526. [Google Scholar] [CrossRef]
- Dong, B.; Miao, J.; Wang, Y.; Luo, W.; Ji, Z.; Lai, H.; Zhang, M.; Cheng, X.; Wang, J.; Fang, Y.; et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun. Biol. 2020, 3, 1–15. [Google Scholar] [CrossRef]
- Kwon, O.-J.; Zhang, L.; Jia, D.; Zhou, Z.; Li, Z.; Haffner, M.; Lee, J.K.; True, L.; Morrissey, C.; Xin, L. De novo induction of lineage plasticity from human prostate luminal epithelial cells by activated AKT1 and c-Myc. Oncogene 2020, 39, 7142–7151. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajabi, H.; Hiraki, M.; Kufe, D. MUC1-C Activates Polycomb Repressive Complexes and Downregulates Tumor Sup-pressor Genes in Human Cancer Cells. Oncogene 2018, 37, 2079–2088. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, A.A.; Vasconcelos, F.F.; Drechsel, D.; Marie, C.; Johnston, C.; Dolle, D.; Bithell, A.; Gillotin, S.; Berg, D.L.V.D.; Ettwiller, L.; et al. Ascl1 Coordinately Regulates Gene Expression and the Chromatin Landscape during Neurogenesis. Cell Rep. 2015, 10, 1544–1556. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor–Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Ng, C.W.; Wamstad, J.A.; Cheng, A.; Thai, K.K.; Fraenkel, E.; Jaenisch, R.; Boyer, L.A. SOX2 Co-Occupies Distal Enhancer Elements with Distinct POU Factors in ESCs and NPCs to Specify Cell State. PLoS Genet. 2013, 9, e1003288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.-C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef]
- Francius, C.; Clotman, F. Dynamic expression of the Onecut transcription factors HNF-6, OC-2 and OC-3 during spinal motor neuron development. Neuroscience 2010, 165, 116–129. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D.; Zhou, T.; Hulsurkar, M.; Ittmann, M.; Shao, L.; Gleave, M.; Li, W. Abstract 186: Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Tumor Biol. 2019, 9, 4080–4082. [Google Scholar] [CrossRef]
- Sang, M.; Hulsurkar, M.; Zhang, X.; Song, H.; Zheng, D.; Zhang, Y.; Li, M.; Xu, J.; Zhang, S.; Ittmann, M.; et al. GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget 2016, 7, 45171–45185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; Macdonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Reina-Campos, M.; Linares, J.F.; Duran, A.; Cordes, T.; L’Hermitte, A.; Badur, M.G.; Bhangoo, M.S.; Thorson, P.K.; Richards, A.; Rooslid, T.; et al. Increased Serine and One-Carbon Pathway Metabolism by PKClambda/Iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 2019, 35, 385–400. [Google Scholar] [CrossRef] [Green Version]
- Faugeroux, V.; Pailler, E.; Oulhen, M.; Deas, O.; Brulle-Soumare, L.; Hervieu, C.; Marty, V.; Alexandrova, K.; Andree, K.C.; Stoecklein, N.H.; et al. Genetic characterization of a unique neuroendocrine transdifferentiation prostate circulating tumor cell-derived eXplant model. Nat. Commun. 2020, 11, 1884. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Romanel, A.; Conteduca, V.; Casiraghi, N.; Sigouros, M.; Franceschini, G.M.; Orlando, F.; Fedrizzi, T.; Ku, S.-Y.; Dann, E.; et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J. Clin. Investig. 2020, 130, 1653–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Wu, C.; Wyatt, A.W.; Lapuk, A.V.; McPherson, A.; McConeghy, B.J.; Bell, R.H.; Anderson, S.; Haegert, A.; Brahmbhatt, S.; Shukin, R.; et al. Integrated genome and transcriptome sequencing identifies a novel form of hybrid and aggressive prostate cancer. J. Pathol. 2012, 227, 53–61. [Google Scholar] [CrossRef]
- Alshalalfa, M.; Liu, Y.; Wyatt, A.W.; Gibb, E.A.; Tsai, H.K.; Erho, N.; Lehrer, J.; Takhar, M.; Ramnarine, V.R.; Collins, C.C.; et al. Characterization of Transcriptomic Signature of Primary Prostate Cancer Analogous to Prostatic Small Cell Neuroendocrine Carcinoma. Int. J. Cancer 2019, 145, 3453–3461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Yu, X. Bioinformatics analyses of publicly available NEPCa datasets. Am. J. Clin. Exp. Urol. 2019, 7, 327–340. [Google Scholar] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; Macdonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 209–224. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Ayala, G.; Dai, H.; Powell, M.; Li, R.; Ding, Y.; Wheeler, T.M.; Shine, D.; Kadmon, D.; Thompson, T.; Miles, B.J.; et al. Cancer-Related Axonogenesis and Neurogenesis in Prostate Cancer. Clin. Cancer Res. 2008, 14, 7593–7603. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, E.M.; Ferretti, P. Considering the evolution of regeneration in the central nervous system. Nat. Rev. Neurosci. 2009, 10, 713–723. [Google Scholar] [CrossRef]
- Nouri, M.; Caradec, J.; Lubik, A.A.; Li, N.; Hollier, B.G.; Takhar, M.; Altimirano-Dimas, M.; Chen, M.; Roshan-Moniri, M.; Butler, M.; et al. Therapy-induced developmental reprogramming of prostate cancer cells and acquired therapy resistance. Oncotarget 2017, 8, 18949–18967. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, B.G.; Bort, A.; Vara-Ciruelos, D.; Díaz-Laviada, I. Androgen Deprivation Induces Reprogramming of Prostate Cancer Cells to Stem-Like Cells. Cells 2020, 9, 1441. [Google Scholar] [CrossRef]
- Grigore, A.D.; Ben-Jacob, E.; Farach-Carson, M.C. Prostate Cancer and Neuroendocrine Differentiation: More Neuronal, Less Endocrine? Front. Oncol. 2015, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D.; et al. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of p53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thaper, D.; Bidnur, S.; Toren, P.; Akamatsu, S.; Bishop, J.L.; Colins, C.; Vahid, S.; Zoubeidi, A. PEG10 is associated with treatment-induced neuroendocrine prostate cancer. J. Mol. Endocrinol. 2019, 63, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Christie, K.J.; Krishnan, A.; Martinez, J.A.; Purdy, K.; Singh, B.; Eaton, S.; Zochodne, D. Enhancing Adult Nerve Regeneration through the Knockdown of Retinoblastoma Protein. Nat. Commun. 2014, 5, 3670. [Google Scholar] [CrossRef]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R.; et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020, 578, 449–454. [Google Scholar] [CrossRef]
- Dominguez, M.H.; Ayoub, A.E.; Rakic, P. POU-III Transcription Factors (Brn1, Brn2, and Oct6) Influence Neurogenesis, Molecular Identity, and Migratory Destination of Upper-Layer Cells of the Cerebral Cortex. Cereb. Cortex 2013, 23, 2632–2643. [Google Scholar] [CrossRef] [Green Version]
- Castro, D.S.; Skowronska-Krawczyk, D.; Armant, O.; Donaldson, I.J.; Parras, C.; Hunt, C.; Critchley, J.A.; Nguyen, L.; Gossler, A.; Gottgens, B.; et al. Proneural bHLH and Brn Proteins Coregulate a Neurogenic Program through Cooperative Binding to a Conserved DNA Motif. Dev. Cell 2006, 11, 831–844. [Google Scholar] [CrossRef]
- Fujii, H.; Hamada, H. A CNS-specific POU transcription factor, Brn-2, is required for establishing mammalian neural cell lineages. Neuron 1993, 11, 1197–1206. [Google Scholar] [CrossRef]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nat. Cell Biol. 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, W.; Jin, H.; Li, T. Brn2 Alone Is Sufficient to Convert Astrocytes into Neural Progenitors and Neurons. Stem Cells Dev. 2018, 27, 736–744. [Google Scholar] [CrossRef]
- Ishii, J.; Sato, H.; Sakaeda, M.; Shishido-Hara, Y.; Hiramatsu, C.; Kamma, H.; Shimoyamada, H.; Fujiwara, M.; Endo, T.; Aoki, I.; et al. POU domain transcription factor BRN2 is crucial for expression of ASCL1, ND1 and neuroendocrine marker molecules and cell growth in small cell lung cancer. Pathol. Int. 2013, 63, 158–168. [Google Scholar] [CrossRef]
- Bhagirath, D.; Yang, T.L.; Tabatabai, Z.L.; Majid, S.; Dahiya, R.; Tanaka, Y.; Saini, S. BRN4 is a Novel Driver of Neuro-endocrine Differentiation in Castration-Resistant Prostate Cancer and is Selectively Released in Extracellular Vesicles with BRNClin. Cancer Res. 2019, 25, 6532–6545. [Google Scholar]
- Suh, H.; Consiglio, A.; Ray, J.; Sawai, T.; D’Amour, K.A.; Gage, F.H. In Vivo Fate Analysis Reveals the Multipotent and Self-Renewal Capacities of Sox2+ Neural Stem Cells in the Adult Hippocampus. Cell Stem Cell 2007, 1, 515–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, R.; Valotta, M.; Ferri, A.L.M.; Latorre, E.; Mariani, J.; Giachino, C.; Lancini, C.; Tosetti, V.; Ottolenghi, S.; Taylor, V.; et al. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 2009, 12, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lujan, E.; Chanda, S.; Ahlenius, H.; Südhof, T.C.; Wernig, M. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. Hierarchical Mechanisms for Direct Reprogramming of Fibroblasts to Neurons. Cell 2013, 155, 621–635. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, L.; Li, Z.; Geng, X.; Li, M.; Tang, Q.; Wu, C.; Lu, Z.-M. SOX2 has dual functions as a regulator in the progression of neuroendocrine prostate cancer. Lab. Investig. 2019, 100, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Vias, M.; Massie, C.E.; East, P.; Scott, H.; Warren, A.; Zhou, Z.; Nikitin, A.Y.; Neal, D.E.; Mills, I.G. Pro-Neural Tran-scription Factors as Cancer Markers. BMC Med. Genom. 2008, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Rapa, I.; Ceppi, P.; Bollito, E.; Rosas, R.; Cappia, S.; Bacillo, E.; Porpiglia, F.; Berruti, A.; Papotti, M.G.; Volante, M. Human ASH1 expression in prostate cancer with neuroendocrine differentiation. Mod. Pathol. 2008, 21, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Ball, D.W. Achaete–Scute homolog-1 and Notch in lung neuroendocrine development and cancer. Cancer Lett. 2004, 204, 159–169. [Google Scholar] [CrossRef]
- Ball, D.W.; Azzoli, C.G.; Baylin, S.B.; Chi, D.; Dou, S.; Donis-Keller, H.; Cumaraswamy, A.; Borges, M.; Nelkin, B.D. Identification of a human achaete-scute homolog highly expressed in neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 1993, 90, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, G.; Dennis, D.; Schuurmans, C. Proneural genes in neocortical development. Neuroscience 2013, 253, 256–273. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Castro, D.S.; Guillemot, F. Proneural genes and the specification of neural cell types. Nat. Rev. Neurosci. 2002, 3, 517–530. [Google Scholar] [CrossRef]
- Castro, D.S.; Martynoga, B.; Parras, C.; Ramesh, V.; Pacary, E.; Johnston, C.; Drechsel, D.; Lebel-Potter, M.; Garcia, L.G.; Hunt, C.; et al. A Novel Function of the Proneural Factor Ascl1 in Progenitor Proliferation Identified by Genome-Wide Characterization of its Targets. Genes Dev. 2011, 25, 930–945. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef]
- Araújo, J.A.D.M.; Hilscher, M.M.; Marques-Coelho, D.; Golbert, D.C.F.; Cornelio, D.A.; De Medeiros, S.R.B.; Leão, R.N.; Costa, M.R. Direct Reprogramming of Adult Human Somatic Stem Cells Into Functional Neurons Using Sox2, Ascl1, and Neurog2. Front. Cell. Neurosci. 2018, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, P.; Lannoy, V.J.; Rousseau, G.G.; Lemaigre, F.P. OC-2, a Novel Mammalian Member of the ONECUT Class of Homeodomain Transcription Factors Whose Function in Liver Partially Overlaps with that of Hepatocyte Nuclear Factor-6. J. Biol. Chem. 1999, 274, 2665–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, J.J.; Martin, G.M.; Chowdhury, R.; Trimarchi, J.M. Onecut1 and Onecut2 Play Critical Roles in the Development of the Mouse Retina. PLoS ONE 2014, 9, e110194. [Google Scholar] [CrossRef] [PubMed]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.G.; Reis-Sobreiro, M.; Huang, W.C.; Huang, F.; Pan, X.; Yanez, A.; Hazelett, D.J.; et al. ONECUT2 is a Targetable Master Regulator of Lethal Prostate Cancer that Suppresses the Androgen Axis. Nat. Med. 2018, 24, 1887–1898. [Google Scholar] [CrossRef]
- Kim, J.; Jin, H.; Zhao, J.C.; Yang, Y.A.; Li, Y.; Yang, X.; Dong, X.; Yu, J. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene 2017, 36, 4072–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenherr, C.J.; Anderson, D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science 1995, 267, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.A.; Tapia-Ramirez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and Its Corepressors Mediate Plasticity of Neuronal Gene Chromatin throughout Neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, T.F.; Hu, G.; Ang, X.L.; Mulligan, P.; Pavlova, N.N.; Liang, A.; Leng, Y.; Maehr, R.; Shi, Y.; Harper, J.W.; et al. SCFbeta-TRCP Controls Oncogenic Transformation and Neural Differentiation through REST Degradation. Nature 2008, 452, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liu, C.; Cui, Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 induces neuroendocrine differentiation (NED) through suppression of RE-1 silencing transcription factor (REST). Prostate 2014, 74, 1086–1094. [Google Scholar] [CrossRef]
- Chen, R.; Li, Y.; Buttyan, R.; Dong, X. Implications of PI3K/AKT inhibition on REST protein stability and neuroendocrine phenotype acquisition in prostate cancer cells. Oncotarget 2017, 8, 84863–84876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borhani, S.; Gartel, A.L. FOXM1: A potential therapeutic target in human solid cancers. Expert Opin. Ther. Targets 2020, 24, 205–217. [Google Scholar] [CrossRef]
- Kelleher, F.C.; O’Sullivan, H. FOXM1 in sarcoma: Role in cell cycle, pluripotency genes and stem cell pathways. Oncotarget 2016, 7, 42792–42804. [Google Scholar] [CrossRef] [Green Version]
- Mosquera, J.M.; Beltran, H.; Park, K.; Macdonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN Gene Amplifications Are Harbingers of Lethal TreatmentRelated Neuroendocrine Prostate Cancer. Neoplasia 2013, 15, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Ton, A.-T.; Singh, K.; Morin, H.; Ban, F.; Leblanc, E.; Lee, J.; Lallous, N.; Cherkasov, A. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 8277. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.A.; Ross-Innes, C.S.; Beraldi, D.; Carroll, J.S.; Balasubramanian, S. Genome-wide mapping of FOXM1 binding reveals co-binding with estrogen receptor alpha in breast cancer cells. Genome Biol. 2013, 14, R6. [Google Scholar] [CrossRef] [Green Version]
- Ketola, K.; Munuganti, R.S.; Davies, A.; Nip, K.M.; Bishop, J.L.; Zoubeidi, A. Targeting Prostate Cancer Subtype 1 by Forkhead Box M1 Pathway Inhibition. Clin. Cancer Res. 2017, 23, 6923–6933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsten, S.L.; Kudo, L.C.; Jackson, R.; Sabatti, C.; Kornblum, H.I.; Geschwind, D.H. Global analysis of gene expression in neural progenitors reveals specific cell-cycle, signaling, and metabolic networks. Dev. Biol. 2003, 261, 165–182. [Google Scholar] [CrossRef] [Green Version]
- Schüller, U.; Zhao, Q.; Godinho, S.A.; Heine, V.M.; Medema, R.H.; Pellman, D.; Rowitch, D.H. Forkhead Transcription Factor FoxM1 Regulates Mitotic Entry and Prevents Spindle Defects in Cerebellar Granule Neuron Precursors. Mol. Cell. Biol. 2007, 27, 8259–8270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, H.; Nakajo, N.; Watanabe, M.; Isoda, M.; Sagata, N. FoxM1-Driven Cell Division is Required for Neuronal Dif-ferentiation in Early Xenopus Embryos. Development 2008, 135, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Zinin, N.; Adameyko, I.; Wilhelm, M.; Fritz, N.; Uhlén, P.; Ernfors, P.; Henriksson, M.A. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. EMBO Rep. 2014, 15, 383–391. [Google Scholar] [CrossRef]
- Adams, R.R.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001, 11, 49–54. [Google Scholar] [CrossRef]
- Lee, E.C.Y.; Frolov, A.; Li, R.; Ayala, G.; Greenberg, N.M. Targeting Aurora Kinases for the Treatment of Prostate Cancer. Cancer Res. 2006, 66, 4996–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chieffi, P.; Cozzolino, L.; Kisslinger, A.; Libertini, S.; Staibano, S.; Mansueto, G.; De Rosa, G.; Villacci, A.; Vitale, M.; Linardopoulos, S.; et al. Aurora B expression directly correlates with prostate cancer malignancy and influence prostate cell proliferation. Prostate 2006, 66, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, J.; Zhou, G.; Mu, D.; Yan, J.; Xing, J.; Yao, Z.; Sheng, H.; Li, D.; Lv, C.; et al. Aurora-A regulates autophagy through the Akt pathway in human prostate cancer. Cancer Biomark. 2017, 19, 27–34. [Google Scholar] [CrossRef]
- Mori, D.; Yamada, M.; Mimori-Kiyosue, Y.; Shirai, Y.; Suzuki, A.; Ohno, S.; Saya, H.; Wynshaw-Boris, A.; Hirotsune, S. An Essential Role of the aPKC-Aurora A-NDEL1 Pathway in Neurite Elongation by Modulation of Microtubule Dynamics. Nat. Cell Biol. 2009, 11, 1057–1068. [Google Scholar] [CrossRef]
- Takitoh, T.; Kumamoto, K.; Wang, C.-C.; Sato, M.; Toba, S.; Wynshaw-Boris, A.; Hirotsune, S. Activation of Aurora-A Is Essential for Neuronal Migration via Modulation of Microtubule Organization. J. Neurosci. 2012, 32, 11050–11066. [Google Scholar] [CrossRef] [Green Version]
- Shlevkov, E.; Basu, H.; Bray, M.-A.; Sun, Z.; Wei, W.; Apaydin, K.; Karhohs, K.; Chen, P.-F.; Smith, J.L.; Wiskow, O.; et al. A High-Content Screen Identifies TPP1 and Aurora B as Regulators of Axonal Mitochondrial Transport. Cell Rep. 2019, 28, 3224–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.A.; Dhele, N.; Cheemadan, S.; Ketkar, A.; Jayandharan, G.R.; Palakodeti, D.; Rampalli, S. Ezh2 mediated H3K27me3 activity facilitates somatic transition during human pluripotent reprogramming. Sci. Rep. 2015, 5, 8229. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Wang, Y.; Hu, Q.; Wu, W.; Wu, Y.; Wei, W.; Han, D.; You, Y.; Lin, N.; Liu, N. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget 2017, 8, 98348–98359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.-W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: A multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 2010, 116, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem. 2015, 58, 5075–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, J.A.; Winston, F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr. Opin. Genet. Dev. 2003, 13, 136–142. [Google Scholar] [CrossRef]
- Seo, S.; Richardson, G.A.; Kroll, K.L. The SWI/SNF chromatin remodeling protein Brg1 is required for vertebrate neurogenesis and mediates transactivation of Ngn and NeuroD. Development 2004, 132, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, S.; Banine, F.; Struve, J.; Xing, R.; Adams, C.; Liu, Y.; Metzger, D.; Chambon, P.; Rao, M.S.; Sherman, L.S. Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev. Biol. 2006, 289, 372–383. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF Nucleosome Remodellers and Cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Cyrta, J.; Augspach, A.; De Filippo, M.R.; Prandi, D.; Thienger, P.; Benelli, M.; Cooley, V.; Bareja, R.; Wilkes, D.; Chae, S.-S.; et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.M.W. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef]
- Farnaby, W.; Koegl, M.; Roy, M.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Davies, A.; Ketola, K.; Zoubeidi, A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocr. Relat. Cancer 2015, 22, R165–R182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.-H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.-E.; et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. eLife 2017, 6, 1309. [Google Scholar] [CrossRef] [Green Version]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Griend, D.J.V.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, T. Study of Androgen Receptor Expression and Neuronal Vulnerability in X-Linked Spinal and Bulbar Muscular Atrophy. Hokkaido Igaku Zasshi 1996, 71, 785–799. [Google Scholar]
- Marron, T.U.; Guerini, V.; Rusmini, P.; Sau, D.; Brevini, T.A.; Martini, L.; Poletti, A. Androgen-Induced Neurite Out-growth is Mediated by Neuritin in Motor Neurones. J. Neurochem. 2005, 92, 10–20. [Google Scholar] [CrossRef]
- Fuller, S.J.; Tan, R.S.; Martins, R.N. Androgens in the Etiology of Alzheimer’s Disease in Aging Men and Possible Therapeutic Interventions. J. Alzheimer’s Dis. 2007, 12, 129–142. [Google Scholar] [CrossRef] [PubMed]
- DonCarlos, L.L.; Garcia-Ovejero, D.; Sarkey, S.; Garcia-Segura, L.M.; Azcoitia, I. Androgen Receptor Immunoreactivity in Forebrain Axons and Dendrites in the Rat. Endocrinology 2003, 144, 3632–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef]
- Kuasne, H.; Barros-Filho, M.C.; Marchi, F.A.; Drigo, S.A.; Scapulatempo-Neto, C.; Faria, E.F.; Rogatto, S.R. Nuclear loss and cytoplasmic expression of androgen receptor in penile carcinomas: Role as a driver event and as a prognosis factor. Virchows Arch. 2018, 473, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Heinlein, C.A.; Chang, C. The Roles of Androgen Receptors and Androgen-Binding Proteins in Nongenomic Androgen Actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Zarif, J.C.; Miranti, C.K. The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell. Signal. 2016, 28, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Efstathiou, E.; Titus, M.; Wen, S.; Hoang, A.; Karlou, M.; Ashe, R.; Tu, S.M.; Aparicio, A.; Troncoso, P.; Mohler, J.; et al. Molecular Characterization of Enzalutamide-treated Bone Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2015, 67, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ci, X.; Hao, J.; Dong, X.; Xue, H.; Wu, R.; Choi, S.Y.; Haegert, A.; Collins, C.C.; Liu, X.; Lin, D.; et al. Conditionally Reprogrammed Cells from Patient-Derived Xenograft to Model Neuroendocrine Prostate Cancer Development. Cells 2020, 9, 1398. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The Beta2-Adrenergic Receptor is a Molecular Switch for Neuroendocrine Transdifferentiation of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 2154–2168. [Google Scholar] [CrossRef] [Green Version]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. Beta-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2015, 4, 375. [Google Scholar] [CrossRef] [Green Version]
- Kulik, G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers 2019, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Bartolomucci, A.; Possenti, R.; Mahata, S.K.; Fischer-Colbrie, R.; Loh, Y.P.; Salton, S.R.J. The Extended Granin Family: Structure, Function, and Biomedical Implications. Endocr. Rev. 2011, 32, 755–797. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative Genomics Analysis Reveals Silencing of Beta-Adrenergic Signaling by Polycomb in Prostate Cancer. Cancer Cell 2007, 12, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, R.A.; Pundavela, J.F.; Biarc, J.; Chalkley, R.J.; Burlingame, A.L.; Hondermarck, H. NGF and ProNGF: Regulation of neuronal and neoplastic responses through receptor signaling. Adv. Biol. Regul. 2015, 58, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, B.; Faulkner, S.; Jobling, P.; Steigler, A.; Blatt, A.; Denham, J.; Hondermarck, H. Tumour Innervation and Neurosignalling in Prostate Cancer. Nat. Rev. Urol. 2020, 17, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Bland, T.; Wang, J.; Yin, L.; Pu, T.; Li, J.; Gao, J.; Lin, T.P.; Gao, A.C.; Wu, B.J. WLS-Wnt Signaling Promotes Neuroendocrine Prostate Cancer. iScience 2021, 24, 101970. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.-Y.H.; Susman, M.W.; Bikoff, J.B.; Ryu, Y.K.; Jonas, A.M.; Hu, L.; Kuruvilla, R.; Greenberg, M.E. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 4044–4051. [Google Scholar] [CrossRef] [Green Version]
- McQuate, A.; Latorre-Esteves, E.; Barria, A. A Wnt/Calcium Signaling Cascade Regulates Neuronal Excitability and Trafficking of NMDARs. Cell Rep. 2017, 21, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-H.; Tarn, W.-Y. Alternative Splicing in Neurogenesis and Brain Development. Front. Mol. Biosci. 2018, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Raj, B.; Irimia, M.; Braunschweig, U.; Sterne-Weiler, T.; O’Hanlon, D.; Lin, Z.-Y.; Chen, G.I.; Easton, L.E.; Ule, J.; Gingras, A.-C.; et al. A Global Regulatory Mechanism for Activating an Exon Network Required for Neurogenesis. Mol. Cell 2014, 56, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Calarco, J.A.; Superina, S.; O’Hanlon, D.; Gabut, M.; Raj, B.; Pan, Q.; Skalska, U.; Clarke, L.; Gelinas, D.; Van Der Kooy, D.; et al. Regulation of Vertebrate Nervous System Alternative Splicing and Development by an SR-Related Protein. Cell 2009, 138, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Kufe, D. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2012, 32, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, H.; Kufe, D. MUC1-C Oncoprotein Integrates a Program of EMT, Epigenetic Reprogramming and Immune Evasion in Human Carcinomas. Biochim. Biophys. Acta Bioenergy 2017, 1868, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C Regulates Lineage Plasticity Driving Progression to Neuroendocrine Prostate Cancer. Nat. Commun. 2020, 11, 338, Erratum in 2020, 11, 1095. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, M.; Yasumizu, Y.; Yamashita, N.; Rajabi, H.; Fushimi, A.; Long, M.D.; Li, W.; Bhattacharya, A.; Ahmad, R.; Oya, M.; et al. Muc1-C Activates the Baf (Mswi/Snf) Complex In Prostate Cancer Stem Cells. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Amit, M.; Na’Ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.A.; Zhou, D.F.; Picker, L.J.; Minty, C.N.; Bargatze, R.F.; Ding, J.F.; Butcher, E.C. A human lymphocyte homing receptor, the Hermes antigen, is related to cartilage proteoglycan core and link proteins. Cell 1989, 56, 1063–1072. [Google Scholar] [CrossRef]
- Li, W.; Cohen, A.; Sun, Y.; Squires, J.; Braas, D.; Graeber, T.G.; Du, L.; Li, G.; Li, Z.; Xu, X.; et al. The Role of CD44 in Glucose Metabolism in Prostatic Small Cell Neuroendocrine Carcinoma. Mol. Cancer Res. 2016, 14, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.A.; Di Sant’Agnese, P.A.; Huang, L.-S.; Xu, H.; Yao, J.L.; Yang, Q.; Liang, S.; Liu, J.; Yu, R.; Cheng, L.; et al. CD44 expression is a feature of prostatic small cell Carcinoma and Distinguishes it from its Mimickers. Hum. Pathol. 2009, 40, 252–258. [Google Scholar] [CrossRef]
- Härkönen, K.; Oikari, S.; Kyykallio, H.; Capra, J.; Hakkola, S.; Ketola, K.; Arasu, U.T.; Daaboul, G.; Malloy, A.; Oliveira, C.; et al. CD44s Assembles Hyaluronan Coat on Filopodia and Extracellular Vesicles and Induces Tumorigenicity of MKN74 Gastric Carcinoma Cells. Cells 2019, 8, 276. [Google Scholar] [CrossRef] [Green Version]
- Dzwonek, J.; Wilczyński, G.M. CD44: Molecular interactions, signaling and functions in the nervous system. Front. Cell. Neurosci. 2015, 9, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketola, K.; Hilvo, M.; Hyötyläinen, T.; Vuoristo, A.; Ruskeepää, A.-L.; Orešič, M.; Kallioniemi, O.; Iljin, K. Salinomycin inhibits prostate cancer growth and migration via induction of oxidative stress. Br. J. Cancer 2012, 106, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Ketola, K.; Vainio, P.; Fey, V.; Kallioniemi, O.; Iljin, K. Monensin Is a Potent Inducer of Oxidative Stress and Inhibitor of Androgen Signaling Leading to Apoptosis in Prostate Cancer Cells. Mol. Cancer Ther. 2010, 9, 3175–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iljin, K.; Ketola, K.; Vainio, P.; Halonen, P.; Kohonen, P.; Fey, V.; Grafstrom, R.C.; Perala, M.; Kallioniemi, O. High-Throughput Cell-Based Screening of 4910 Known Drugs and Drug-Like Small Molecules Identifies Disulfiram as an Inhibitor of Prostate Cancer Cell Growth. Clin. Cancer Res. 2009, 15, 6070–6078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketola, K.; Kallioniemi, O.; Iljin, K. Chemical Biology Drug Sensitivity Screen Identifies Sunitinib as Synergistic Agent with Disulfiram in Prostate Cancer Cells. PLoS ONE 2012, 7, e51470. [Google Scholar] [CrossRef]
- Amador-Arjona, A.; Cimadamore, F.; Huang, C.-T.; Wright, R.; Lewis, S.; Gage, F.H.; Terskikh, A.V. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E1936–E1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, A.L.M.; Cavallaro, M.; Braida, D.; Di Cristofano, A.; Canta, A.; Vezzani, A.; Ottolenghi, S.; Pandolfi, P.P.; Sala, M.; DeBiasi, S.; et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 2004, 131, 3805–3819. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Van Schaijik, B.; Davis, P.F.; Wickremesekera, A.C.; Tan, S.T.; Itinteang, T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J. Clin. Pathol. 2017, 71, 88–91. [Google Scholar] [CrossRef]
- Kretschmer, A.; Zhang, F.; Somasekharan, S.P.; Tse, C.; Leachman, L.; Gleave, A.; Li, B.; Asmaro, I.; Huang, T.; Kotula, L.; et al. Stress-induced tunneling nanotubes support treatment adaptation in prostate cancer. Sci. Rep. 2019, 9, 7826. [Google Scholar] [CrossRef]
- Rustom, A. The missing link: Does tunnelling nanotube-based supercellularity provide a new understanding of chronic and lifestyle diseases? Open Biol. 2016, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibinger, M.; Muller, A.M.S.; Gobrecht, P.; Diekmann, H.; Andreadaki, A.; Fischer, D. Interleukin-6 contributes to CNS axon regeneration upon inflammatory stimulation. Cell Death Dis. 2013, 4, e609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.T.; Kwon, S.J.; Lee, J.-H.; Jeon, S.S.; Jang, K.T.; Choi, H.Y.; Lee, H.M.; Kim, W.-J.; Lee, D.H.; Kim, I.Y. Macrophages induce neuroendocrine differentiation of prostate cancer cells via BMP6-IL6 Loop. Prostate 2011, 71, 1525–1537. [Google Scholar] [CrossRef]
- Burchardt, T.; Burchardt, M.; Chen, M.W.; Cao, Y.; de la Taille, A.; Shabsigh, A.; Hayek, O.; Dorai, T.; Buttyan, R. Transdifferentiation of Prostate Cancer Cells to a Neuroendocrine Cell Phenotype in Vitro and in Vivo. J. Urol. 1999, 162, 1800–1805. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.-P.; Firlej, V.; Allory, Y.; Roméo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nat. Cell Biol. 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Yin, L.; Hu, P.; Shi, X.; Qian, W.; Zhau, H.E.; Pandol, S.J.; Lewis, M.S.; Chung, L.W.K.; Wang, R. Cancer Cell’s Neuro-endocrine Feature can be Acquired through Cell-Cell Fusion during Cancer-Neural Stem Cell Interaction. Sci. Rep. 2020, 10, 1216. [Google Scholar] [CrossRef] [Green Version]
- Sroka, I.C.; Chopra, H.; Das, L.; Gard, J.M.; Nagle, R.B.; Cress, A.E. Schwann Cells Increase Prostate and Pancreatic Tumor Cell Invasion Using Laminin Binding A6 Integrin. J. Cell. Biochem. 2016, 117, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes-Villagrana, R.D.; Albores-Garcia, D.; Cervantes-Villagrana, A.R.; Garcia-Acevez, S.J. Tumor-Induced Neurogenesis and Immune Evasion as Targets of Innovative Anti-Cancer Therapies. Signal Transduct. Target Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Zhu, S.; Tian, H.; Niu, X.; Wang, J.; Li, X.; Jiang, N.; Wen, S.; Chen, X.; Ren, S.; Xu, C.; et al. Neurotensin and its receptors mediate neuroendocrine transdifferentiation in prostate cancer. Oncogene 2019, 38, 4875–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, D.; Hautot, C.; Mehio, M.; Jeandel, L.; Courel, M.; Voisin, T.; Couvineau, A.; Gobet, F.; Leprince, J.; Pfister, C.; et al. The orexin type 1 receptor is overexpressed in advanced prostate cancer with a neuroendocrine differentiation, and mediates apoptosis. Eur. J. Cancer 2014, 50, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cañas, I.; Juarranz, M.G.; Collado, B.; Rodríguez-Henche, N.; Chiloeches, A.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide induces neuroendocrine differentiation in the LNCaP prostate cancer cell line through PKA, ERK, and PI3K. Prostate 2004, 63, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Alshalalfa, M.; Nguyen, P.L.; Beltran, H.; Chen, W.S.; Davicioni, E.; Zhao, S.G.; Rebbeck, T.R.; Schaeffer, E.M.; Lotan, T.L.; Feng, F.Y.; et al. Transcriptomic and Clinical Characterization of Neuropeptide Y Expression in Localized and Metastatic Prostate Cancer: Identification of Novel Prostate Cancer Subtype with Clinical Implications. Eur. Urol. Oncol. 2019, 2, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Ketola, K.; Viitala, M.; Kohonen, P.; Fey, V.; Culig, Z.; Kallioniemi, O.; Iljin, K. High-throughput cell-based compound screen identifies pinosylvin methyl ether and tanshinone IIA as inhibitors of castration-resistant prostate cancer. J. Mol. Biochem. 2016, 5, 12–22. [Google Scholar] [PubMed]
- Cole, S.W.; Sood, A.K. Molecular Pathways: Beta-Adrenergic Signaling in Cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Powe, D.G.; Entschladen, F. Targeted Therapies: Using Beta-Blockers to Inhibit Breast Cancer Progression. Nat. Rev. Clin. Oncol. 2011, 8, 511–512. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Agent | Target | Clinical Status |
|---|---|---|
| Cancer stem Cell Inhibitors | ||
| Disulfiram | ALDH1A1 inhibitor | Phase Ib |
| Rovalpituzumab tesirine (Rova-T) | DLL3 targeting agent | Phase I |
| MYCN and AURKA inhibitors | ||
| MLN8237 | N-Myc inhibition | Phase II |
| Alisertib (MLN8237) | AURKA inhibition | Phase II |
| BET inhibitors | ||
| ZEN003694 | BET protein inhibition | Phase I |
| GS-5829 | BET protein inhibition | Phase I and II |
| Epigenetic modulators/EZH2 inhibitors | ||
| GSK2816126 | EZH2 activity inhibition | Phase I |
| Tazemetostat (EPZ-6438) | EZH2 activity inhibition | Phase II |
| CPI-1205 | EZH2 activity inhibition | Phase Ib and II |
| LSD1 inhibitor | ||
| INCB059872 | LSD1 inhibition | Phase I and II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaarijärvi, R.; Kaljunen, H.; Ketola, K. Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression. Cancers 2021, 13, 692. https://doi.org/10.3390/cancers13040692
Kaarijärvi R, Kaljunen H, Ketola K. Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression. Cancers. 2021; 13(4):692. https://doi.org/10.3390/cancers13040692
Chicago/Turabian StyleKaarijärvi, Roosa, Heidi Kaljunen, and Kirsi Ketola. 2021. "Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression" Cancers 13, no. 4: 692. https://doi.org/10.3390/cancers13040692