
Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential

 , ,
, ,  ,
,  ,
,
Abstract
:Simple Summary
Abstract
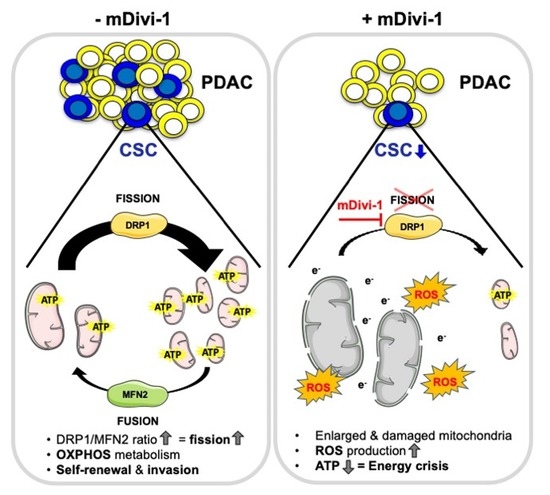
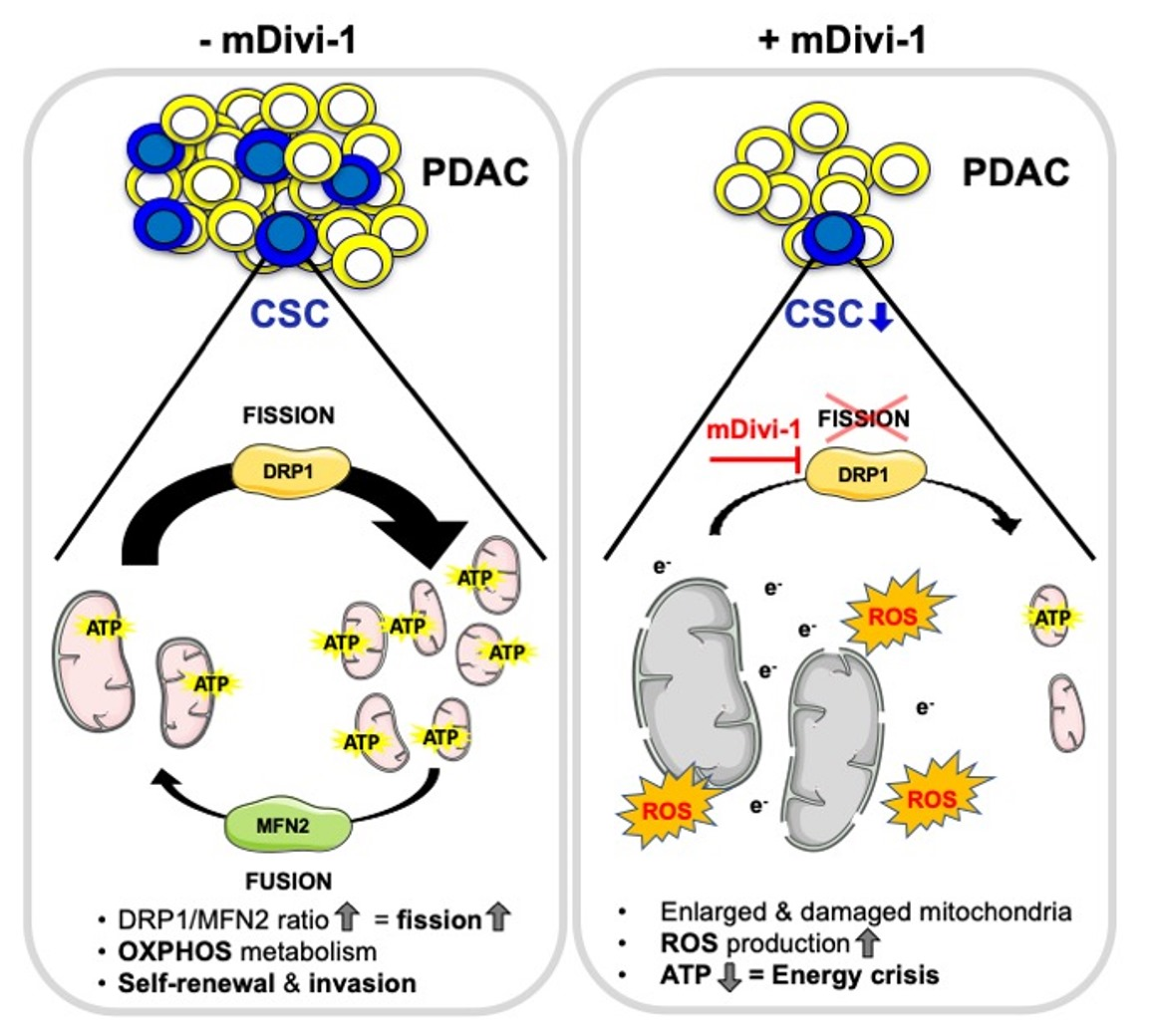
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Patient-Derived Xenografts
2.2. Treatments
2.3. Human Data Analysis
2.4. Transmission Electron Microscopy (TEM)
2.5. Immunoblots
2.6. RNA Extraction and Quantitative Reverse Transcription Polymerase Chain Reaction (RTqPCR)
2.7. Proliferation Assay
2.8. Cytotoxicity Assay
2.9. Flow Cytometry Analysis and Sorting
2.10. XF Extracellular Flux Analyzer Experiments
2.11. ATP Measurement
2.12. Sphere Formation Assay
2.13. Colony Formation Assay
2.14. Invasion Assay
2.15. Wound Healing Assay
2.16. In vivo Extreme Limiting Dilution Assay (ELDA)
2.17. Statistical Analysis
3. Results
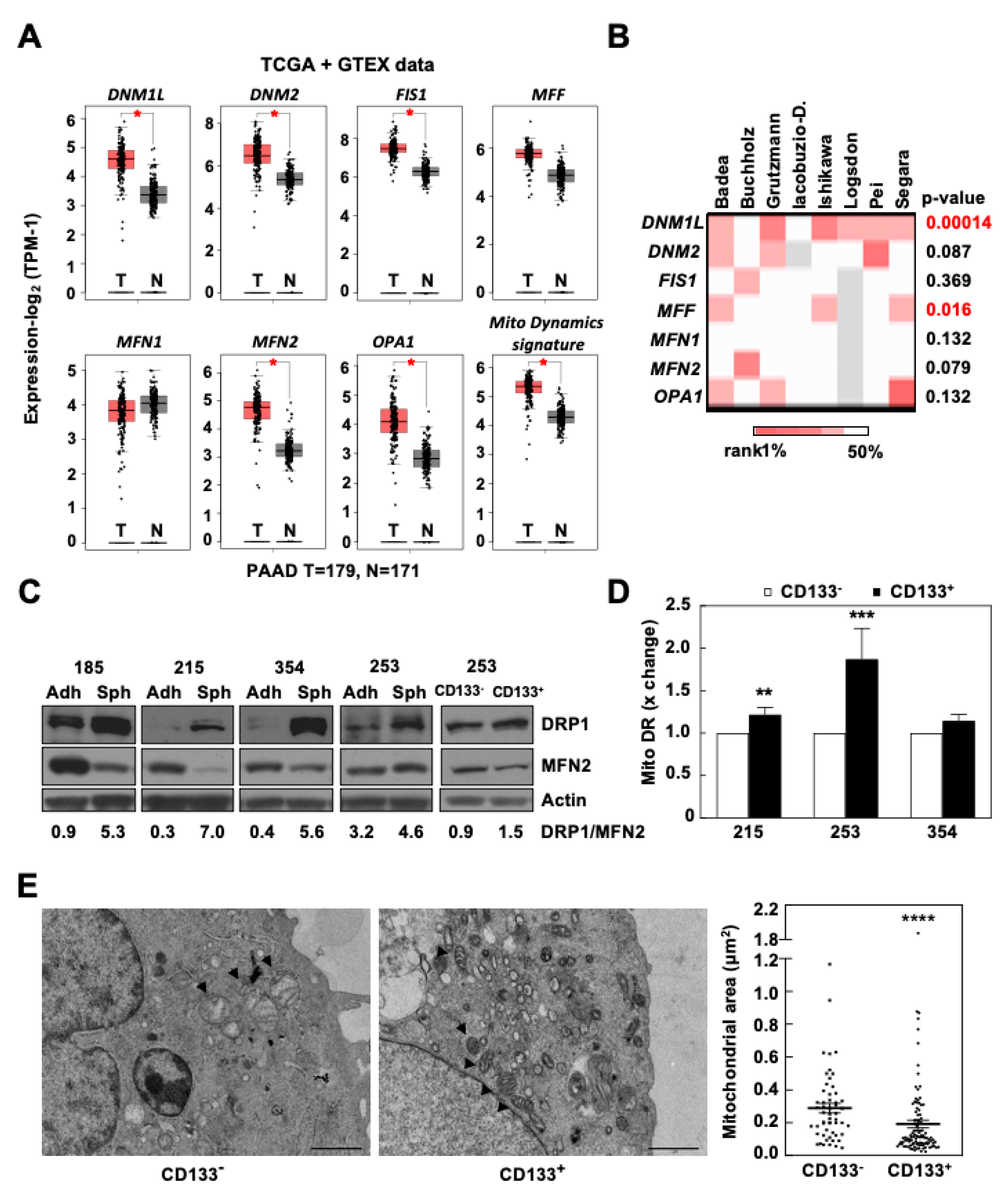
3.1. Mitochondrial Fission Is Associated with Stemness and Epithelial-to-Mesenchymal Transition in Human PDAC
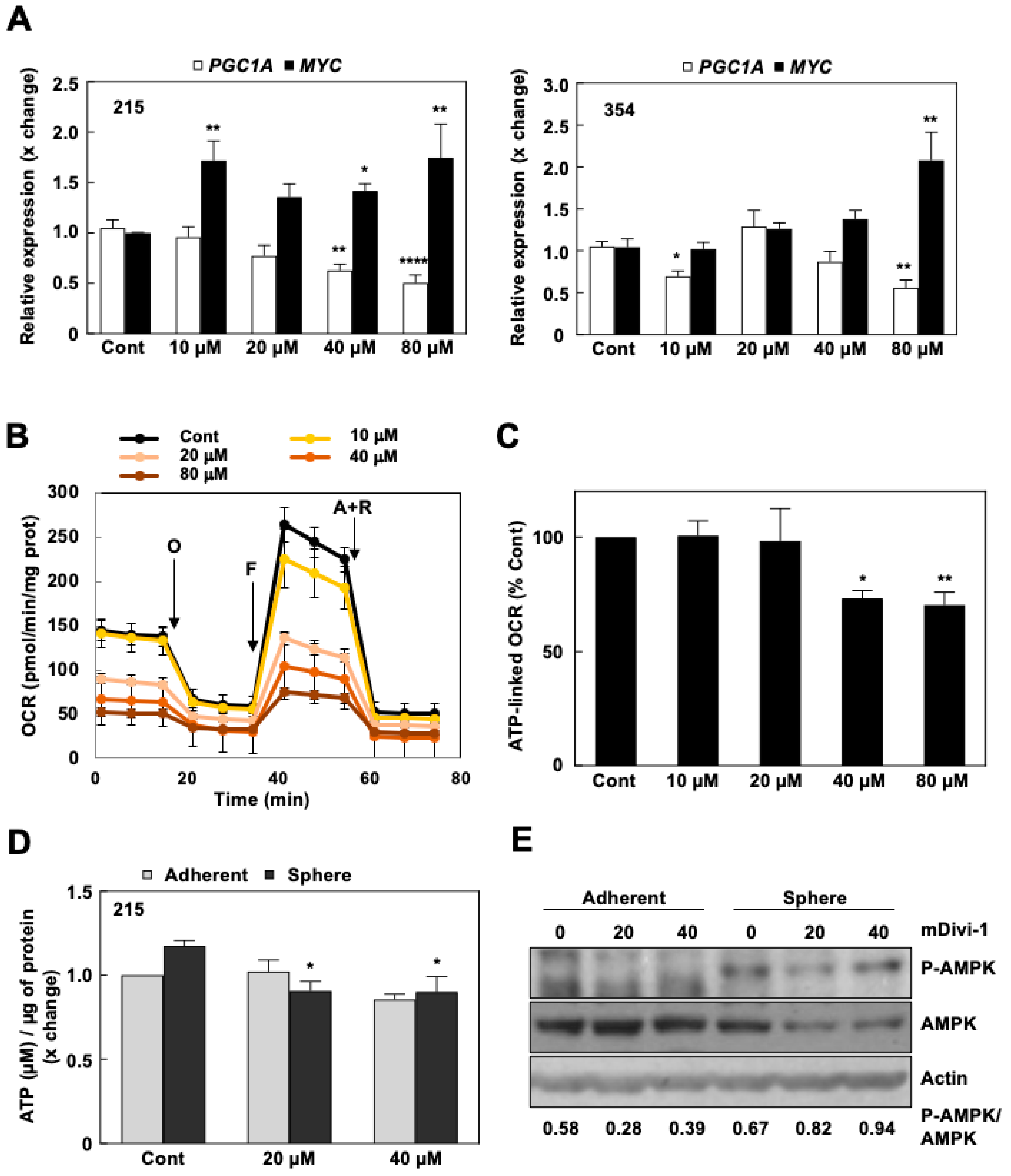
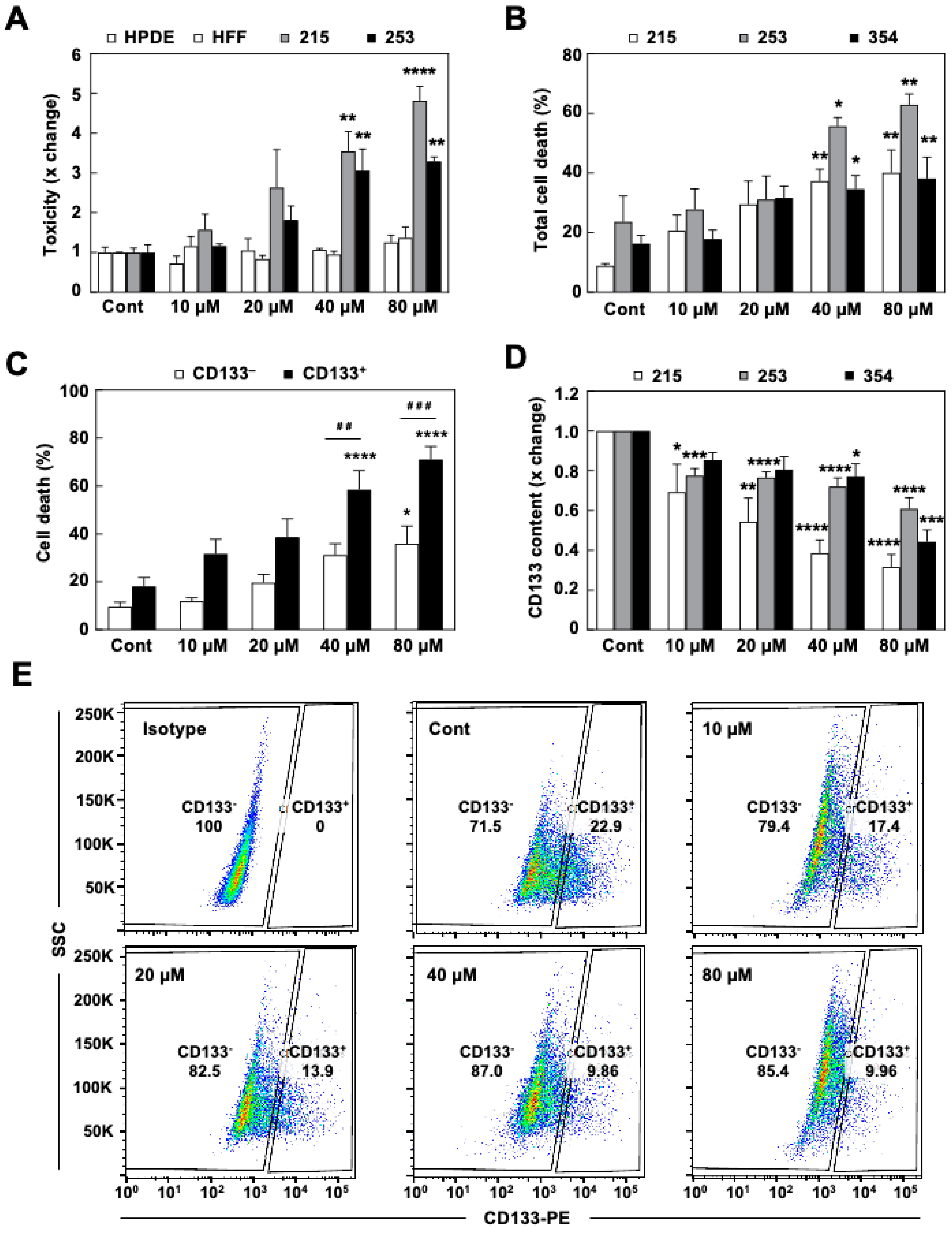
3.2. The DRP1 Inhibitor mDivi-1 Induces Apoptosis in Primary Pancreatic Cancer Cells, Especially Affecting the CD133+ Subpopulation
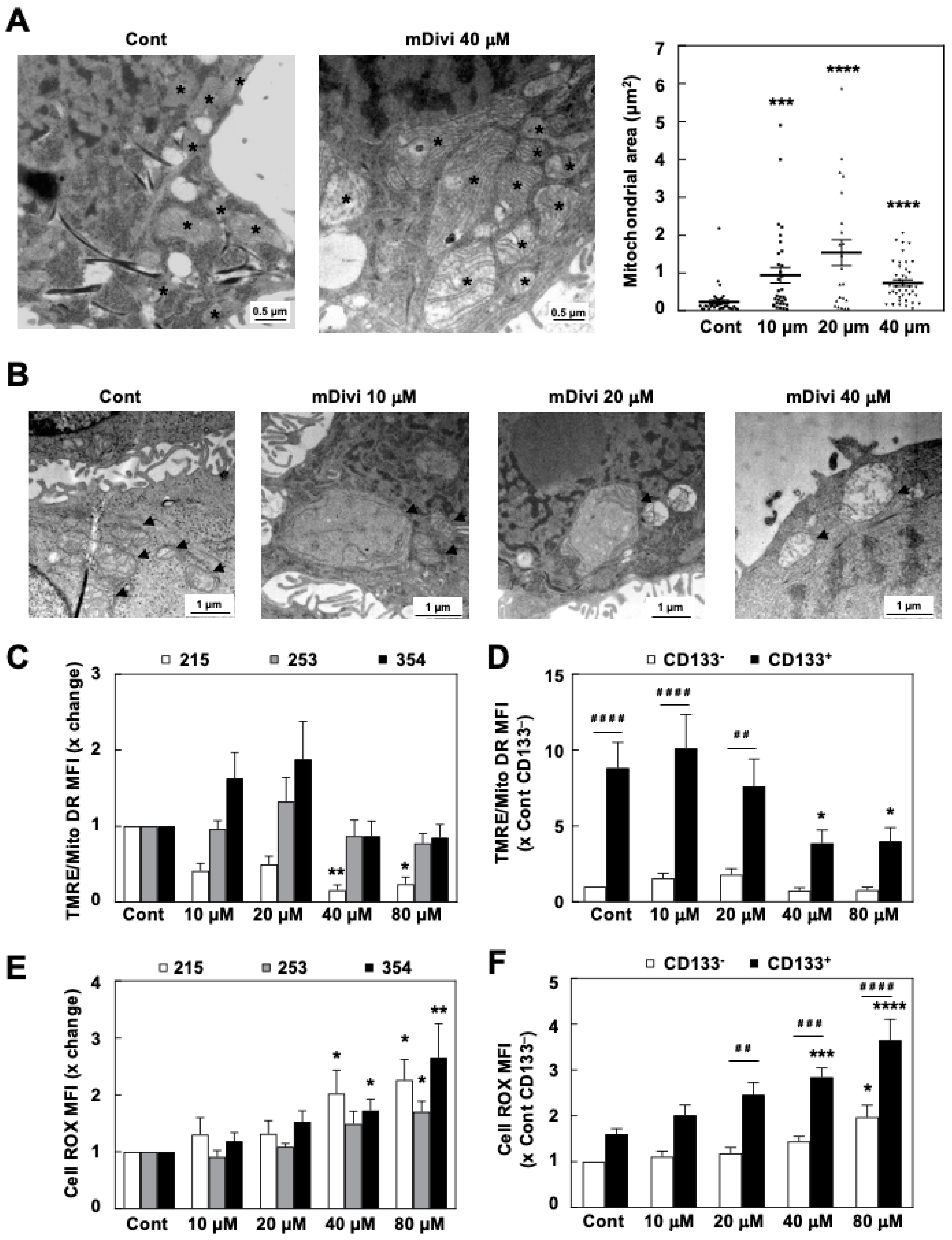
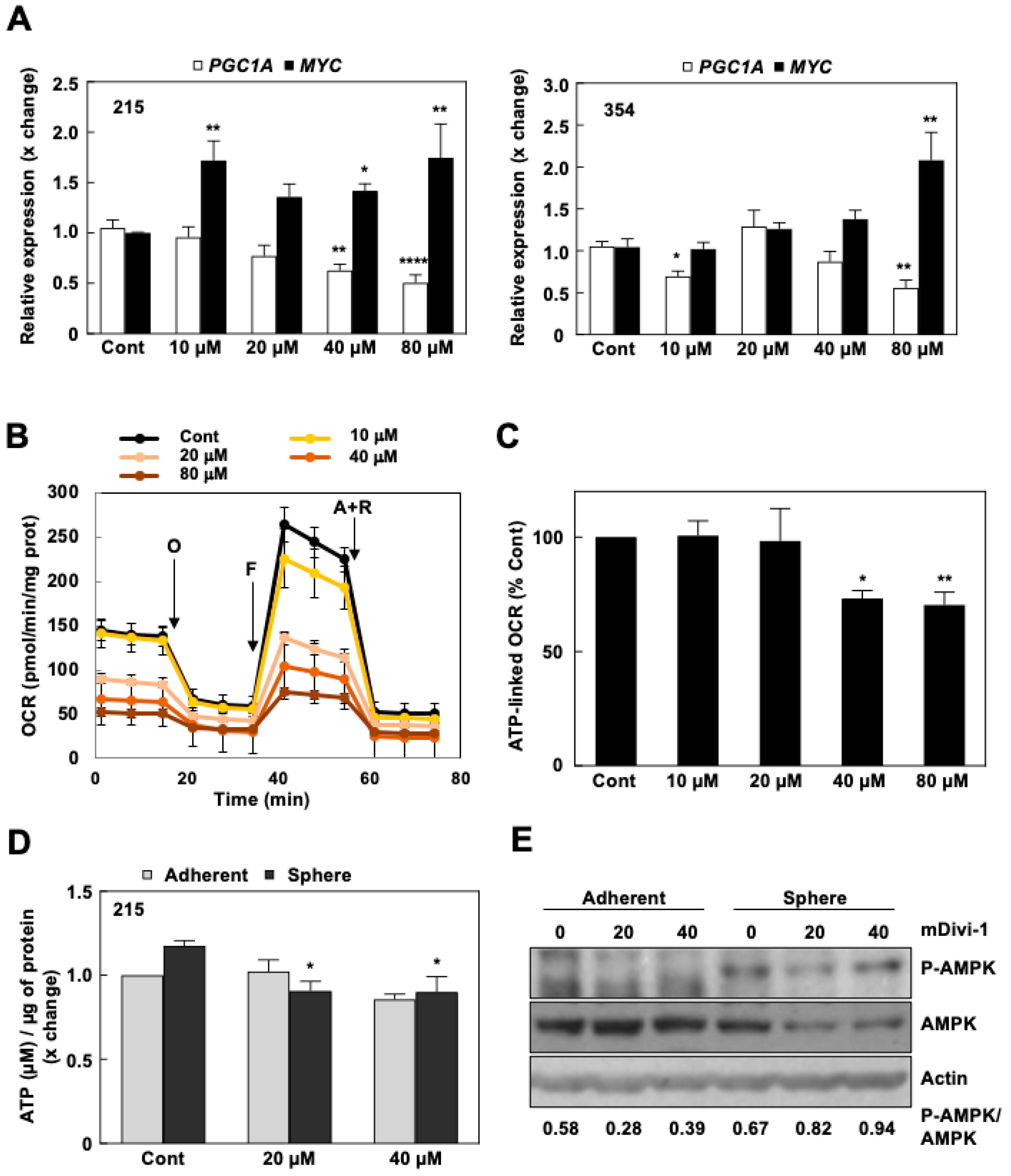
3.3. mDivi-1 Treatment Disrupts Mitochondrial Function
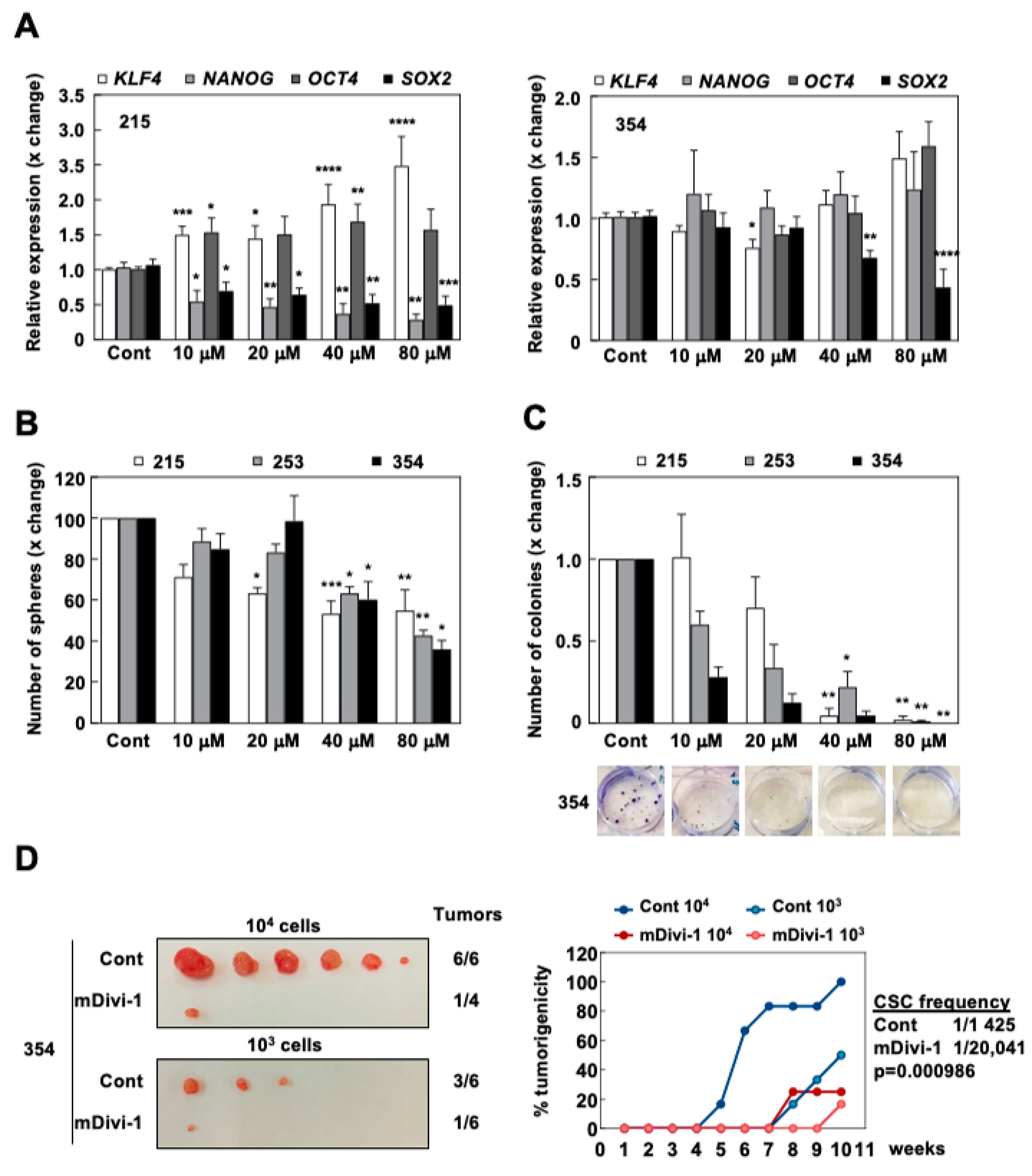
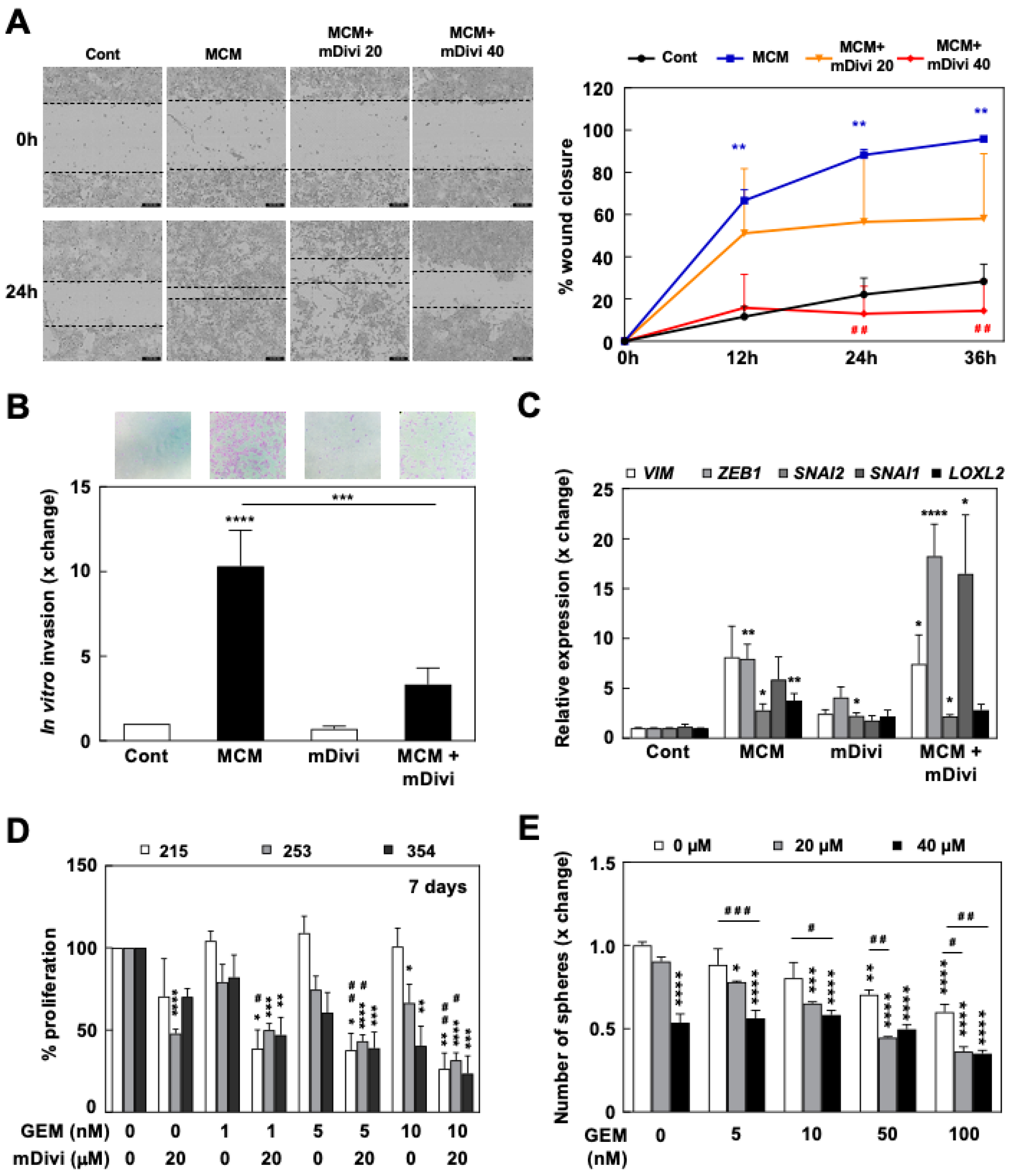
3.4. mDivi-1 Treatment Blocks CSC Functionality
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study. Lancet 2018, 392, 1923–1994. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Alcala, S.; Usachov, V.; Hermann, P.C.; Sainz, J.B. The ever-changing landscape of pancreatic cancer stem cells. Pancreatolgy 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Lorenzo, I.; Dorado, J.; Lonardo, E.; Alcala, S.; Serrano, A.G.; Clausell-Tormos, J.; Cioffi, M.; Megias, D.; Zagorac, S.; Balic, A.; et al. Intracellular autofluorescence: A biomarker for epithelial cancer stem cells. Nat. Methods 2014, 11, 1161–1169. [Google Scholar] [CrossRef]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y.; Miranda-Lorenzo, I.; Lonardo, E.; Dorado, J.; Vieira, C.R.; Ramirez, J.C.; Hidalgo, M.; Aicher, A.; et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut 2015, 64, 1936–1948. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [Green Version]
- Jagust, P.; Alcalá, S.; Jr, B.S.; Heeschen, C.; Sancho, P. Glutathione metabolism is essential for self-renewal and chemoresistance of pancreatic cancer stem cells. World J. Stem Cells 2020, 12, 1410–1428. [Google Scholar] [CrossRef] [PubMed]
- Alcalá, S.; Sancho, P.; Martinelli, P.; Navarro, D.; Pedrero, C.; Martín-Hijano, L.; Valle, S.; Earl, J.; Rodríguez-Serrano, M.; Ruiz-Cañas, L.; et al. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat. Commun. 2020, 11, 2682. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Jagust, P.; De Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859.e5. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; Lucas, F.A.S.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 4, 126915. [Google Scholar] [CrossRef]
- Mueller, M.; Hermann, P.C.; Witthauer, J.; Rubio–Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined Targeted Treatment to Eliminate Tumorigenic Cancer Stem Cells in Human Pancreatic Cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Valle, S.; Alcalá, S.; Martin-Hijano, L.; Cabezas-Sáinz, P.; Navarro, D.; Muñoz, E.R.; Yuste, L.; Tiwary, K.; Walter, K.; Ruiz-Cañas, L.; et al. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Sainz, B.; Martín, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 Is a Critical Microenvironmental Factor for Pancreatic Cancer Stem Cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef] [Green Version]
- Sainz, J.B.; Alcala, S.; Garcia, E.; Sanchez-Ripoll, Y.; Azevedo, M.M.; Cioffi, M.; Tatari, M.; Miranda-Lorenzo, I.; Hidalgo, M.; Gomez-Lopez, G.; et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015, 64, 1921–1935. [Google Scholar] [CrossRef] [Green Version]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [Green Version]
- Bokil, A.; Sancho, P. Mitochondrial determinants of chemoresistance. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [Green Version]
- Son, M.J.; Choi, H.; Han, Y.-M.; Cho, Y.S. Unveiling the critical role of REX1 in the regulation of human stem cell pluripotency. STEM CELLS 2013, 31, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; Maclaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.-E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects-Cells From Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nat. Cell Biol. 2001, 414, 807–812. [Google Scholar] [CrossRef]
- Lei, X.G.; Vatamaniuk, M. Two Tales of Antioxidant Enzymes onβCells and Diabetes. Antioxidants Redox Signal. 2011, 14, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pagès, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254–13275. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Da-Silva, A.; Valacca, C.; Rios, E.; Pópulo, H.; Soares, P.; Sobrinho-Simões, M.; Scorrano, L.; Máximo, V.; Campello, S. Mitochondrial Dynamics Protein Drp1 Is Overexpressed in Oncocytic Thyroid Tumors and Regulates Cancer Cell Migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Tsuyoshi, H.; Orisaka, M.; Shieh, D.-B.; Yoshida, Y.; Tsang, B.K. Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann. N. Y. Acad. Sci. 2015, 1350, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-J.; Yang, Z.-J.; Jiang, L.-P.; Wei, Y.-F.; Liao, M.-F.; Qian, Y.; Li, Y.; Huang, X.; Wang, J.-B.; Xin, H.; et al. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int. J. Oncol. 2014, 46, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Wang, J.; Roginskaya, V.; McDermott, L.A.; Edwards, R.P.; Stolz, D.B.; Llambi, F.; Green, D.R.; Van Houten, B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.; Gallo, G. To mdivi-1 or not to mdivi-1: Is that the question? Dev. Neurobiol. 2017, 77, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Rosdah, A.A.; Holien, J.K.; Delbridge, L.M.D.; Dusting, G.J.; Lim, S.Y. Mitochondrial fission—A drug target for cytoprotection or cytodestruction? Pharmacol. Res. Perspect. 2016, 4, e00235. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| HPRT | TGACCTTGATTTATTTTGCATACC | CGAGCAAGACGTTCAGTCCT |
| C-MYC | CCCGCTTCTCTGAAAGGCTCTC | CTCTGCTGCTGCTGCTGGTAG |
| PGC-1A | TGACTGGCGTCATTCAGGAG | CCAGAGCAGCACACTCGAT |
| NANOG | AGAACTCTCCAACATCCTGAACCT | TGCCACCTCTTAGATTTCATTCTCT |
| OCT3/4 | CTTGCTGCAGAAGTGGGTGGAGGAA | CTGCAGTGTGGGTTTCGGGCA |
| SOX2 | AGAACCCCAAGATGCACAAC | CGGGGCCGGTATTTATAATC |
| KLF4 | ACCCACACAGGTGAGAAACC | ATGTGTAAGGCGAGGTGGTC |
| LOXL2 | GGCACCGTGTTGCGATGACGA | GCTGCAAGGGTCGCCTCGTT |
| SNAIL | GCTCCTTCGTCCTTCTCCTC | TGACATCTGAGTGGGTCTGG |
| SLUG | GTGTTTGCAAGATCTGCGGC | TTCTCCCCCGTGTGAGTTCT |
| VIM | GACAATGCGTCTCTGGCACGTCTT | TCCTCCGCCTCCTGCAGGTTCTT |
| ZEB1 | GATGATGAATGCGAGTCAGATGC | CTGGTCCTCTTCAGGTGCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Courtois, S.; de Luxán-Delgado, B.; Penin-Peyta, L.; Royo-García, A.; Parejo-Alonso, B.; Jagust, P.; Alcalá, S.; Rubiolo, J.A.; Sánchez, L.; Sainz, B., Jr.; et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers 2021, 13, 698. https://doi.org/10.3390/cancers13040698
Courtois S, de Luxán-Delgado B, Penin-Peyta L, Royo-García A, Parejo-Alonso B, Jagust P, Alcalá S, Rubiolo JA, Sánchez L, Sainz B Jr., et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers. 2021; 13(4):698. https://doi.org/10.3390/cancers13040698
Chicago/Turabian StyleCourtois, Sarah, Beatriz de Luxán-Delgado, Laure Penin-Peyta, Alba Royo-García, Beatriz Parejo-Alonso, Petra Jagust, Sonia Alcalá, Juan A. Rubiolo, Laura Sánchez, Bruno Sainz, Jr., and et al. 2021. "Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential" Cancers 13, no. 4: 698. https://doi.org/10.3390/cancers13040698
APA StyleCourtois, S., de Luxán-Delgado, B., Penin-Peyta, L., Royo-García, A., Parejo-Alonso, B., Jagust, P., Alcalá, S., Rubiolo, J. A., Sánchez, L., Sainz, B., Jr., Heeschen, C., & Sancho, P. (2021). Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers, 13(4), 698. https://doi.org/10.3390/cancers13040698