
Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology



Abstract
Simple Summary
Abstract
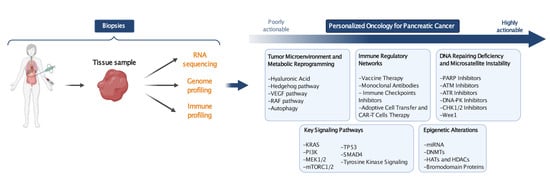
1. Introduction
2. Methods
3. Targeting DNA Repairing Deficiency and Microsatellite Instability
3.1. PARP Inhibitors
3.2. ATM Inhibition
3.3. ATR Inhibition
3.4. DNA-PK Inhibition
3.5. CHK1/2 Inhibition
3.6. Wee1
4. Targeting Epigenetic Alterations
4.1. miRNA
4.2. DNMTs
4.3. HATs and HDACs
4.4. Bromodomain Proteins
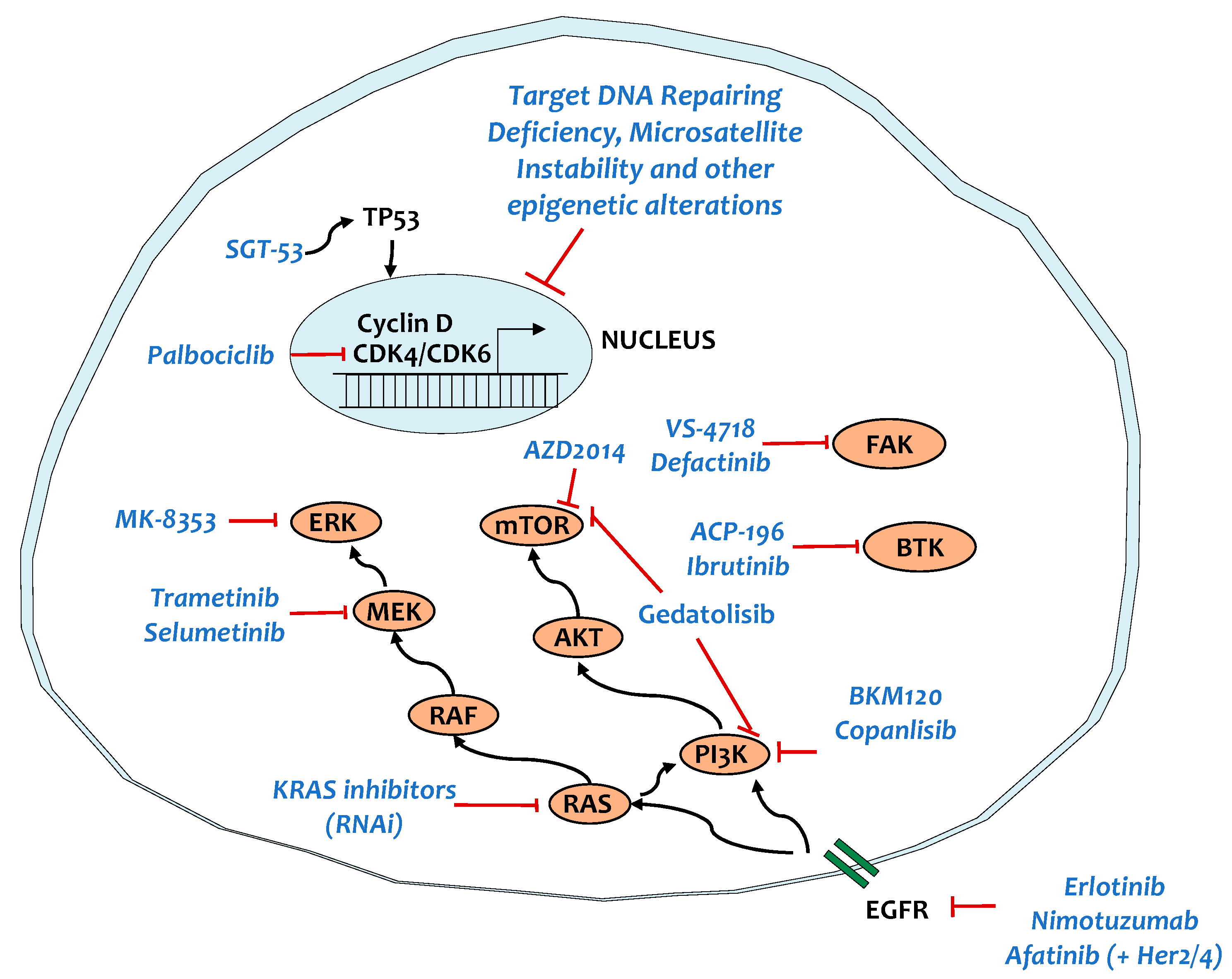
5. Targeting Key Signaling Pathways
5.1. KRAS and Main Targets
5.2. TP53
5.3. SMAD4
5.4. Tyrosine Kinase Signaling
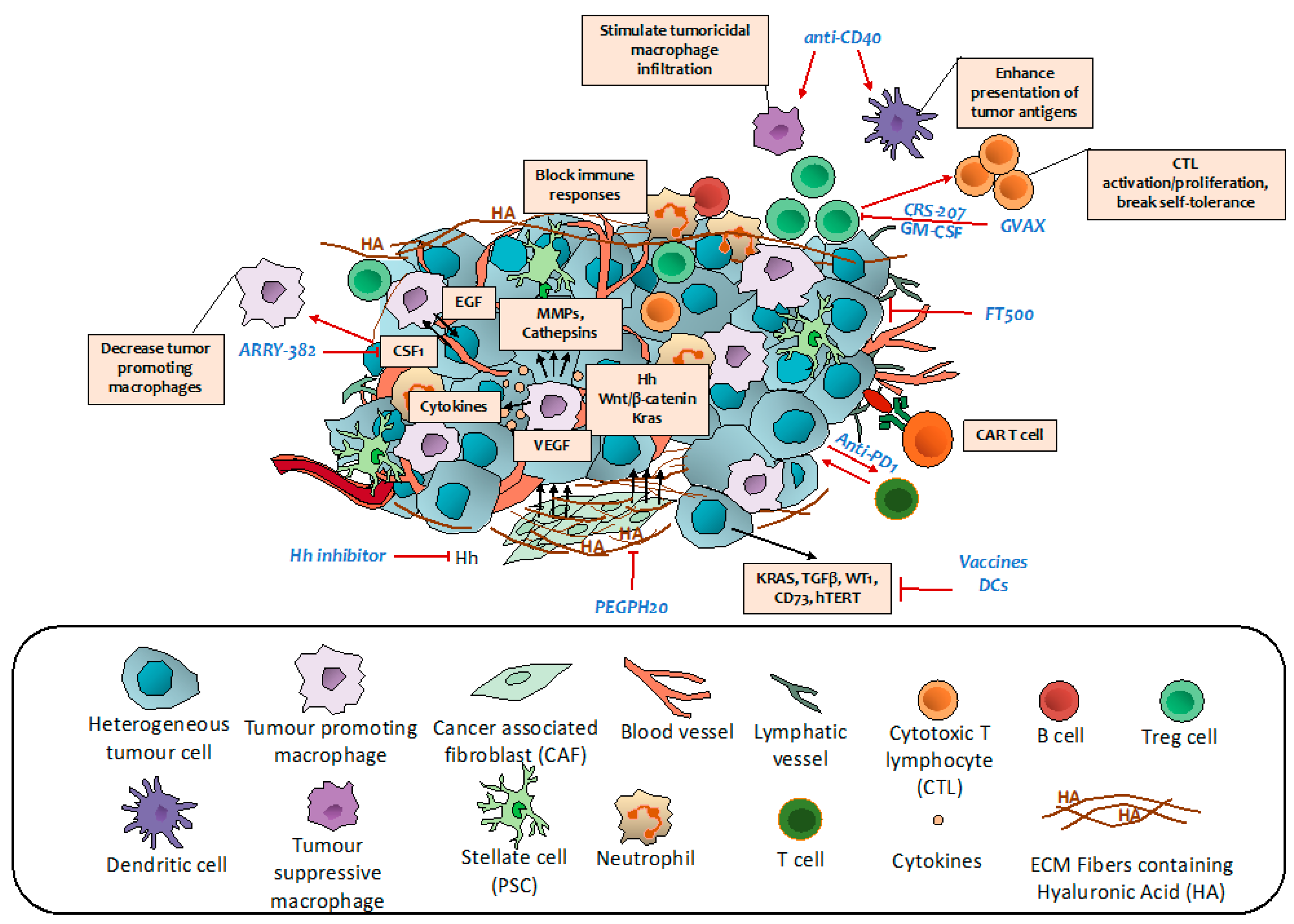
6. Targeting the Tumor Microenvironment and Related Metabolic Reprogramming
7. Targeting Immune Regulatory Networks
7.1. Vaccine Therapy
7.2. Monoclonal Antibodies
7.3. Immune Checkpoints Inhibitors
7.4. Adoptive Cell Transfer Therapy
7.5. Other Immune Regulators
8. The Road to Personalized Oncology
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Sohal, D.P.S.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3217–3230. [Google Scholar] [CrossRef]
- Balaban, E.P.; Mangu, P.B.; Yee, N.S. Locally Advanced Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Summary. J. Oncol. Pract. 2017, 13, 265–269. [Google Scholar] [CrossRef]
- Khorana, A.A.; Mangu, P.B.; Berlin, J.; Engebretson, A.; Hong, T.S.; Maitra, A.; Mohile, S.G.; Mumber, M.; Schulick, R.; Shapiro, M.; et al. Potentially Curable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 2324–2328. [Google Scholar] [CrossRef]
- Khorana, A.A.; McKernin, S.E.; Berlin, J.; Hong, T.S.; Maitra, A.; Moravek, C.; Mumber, M.; Schulick, R.; Zeh, H.J.; Katz, M.H.G. Potentially Curable Pancreatic Adenocarcinoma: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2019, 37, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Hall, W.A.; Goodman, K.A. Radiation therapy for pancreatic adenocarcinoma, a treatment option that must be considered in the management of a devastating malignancy. Radiat Oncol. 2019, 14, 114. [Google Scholar] [CrossRef]
- Berrington de Gonzalez, A.; Sweetland, S.; Spencer, E. A meta-analysis of obesity and the risk of pancreatic cancer. Br. J. Cancer 2003, 89, 519–523. [Google Scholar] [CrossRef]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef]
- Elena, J.W.; Steplowski, E.; Yu, K.; Hartge, P.; Tobias, G.S.; Brotzman, M.J.; Chanock, S.J.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; et al. Diabetes and risk of pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium. Cancer Causes Control. 2013, 24, 13–25. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; la Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.M.; Fernandez-Zapico, M.E. Toward personalized TGFβ inhibition for pancreatic cancer. EMBO Mol. Med. 2019, 11, e11414. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Maitra, A. Subtyping Pancreatic Cancer. Cancer Cell 2015, 28, 411–413. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Feldmann, G.; Zander, T.; Reinhardt, H.C. Targeting Defects in the Cellular DNA Damage Response for the Treatment of Pancreatic Ductal Adenocarcinoma. Oncol. Res. Treat. 2018, 41, 619–625. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; de Almeida, F.K.; Simoes, C.B.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A., 3rd; de Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, H., III. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef]
- Kamphues, C.; Bova, R.; Bahra, M.; Klauschen, F.; Muckenhuber, A.; Sinn, B.V.; Warth, A.; Goeppert, B.; Endris, V.; Neuhaus, P.; et al. Ataxia-telangiectasia-mutated protein kinase levels stratify patients with pancreatic adenocarcinoma into prognostic subgroups with loss being a strong indicator of poor survival. Pancreas 2015, 44, 296–301. [Google Scholar] [CrossRef]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Güthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef]
- Gout, J.; Perkhofer, L.; Morawe, M.; Arnold, F.; Ihle, M.; Biber, S.; Lange, S.; Roger, E.; Kraus, J.M.; Stifter, K.; et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Sundar, R.; Brown, J.; Russo, A.I.; Yap, T.A. Targeting ATR in cancer medicine. Curr. Probl. Cancer 2017, 41, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.B.; Fornari, C.; Yates, J.W.T.; Fernandez, S.B.d.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef]
- O’Carrigan, B.; de Miguel Luken, M.; Papadatos-Pastos, D.; Brown, J.; Tunariu, N.; Perez-Lopez, R.; Ganegoda, M.; Riisnaes, R.; Figueiredo, I.; Carreira, S.; et al. Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J. Clin. Oncol. 2016, 34, 2504. [Google Scholar] [CrossRef]
- Plummer, E.R.; Dean, E.J.; Evans, T.R.J.; Greystoke, A.; Herbschleb, K.; Ranson, M.; Brown, J.; Zhang, Y.; Karan, S.; Pollard, J.; et al. Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792). J. Clin. Oncol. 2016, 34 (Suppl. S15), 2513. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.d.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Willers, H.; Dahm-Daphi, J.; Powell, S.N. Repair of radiation damage to DNA. Br. J. Cancer 2004, 90, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Lees-Miller, S.P.; Godbout, R.; Chan, D.W.; Weinfeld, M.; Day, R.S., III; Barron, G.M.; Allalunis-Turner, J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science 1995, 267, 1183–1185. [Google Scholar] [CrossRef]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef]
- Van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sorensen, M.; Nielsen, D.; Schoffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; el Bawab, S.; et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Esaki, T.; Hirai, F.; Arita, S.; Nosaki, K.; Makiyama, A.; Kometani, T.; Fujimoto, C.; Hamatake, M.; Takeoka, H.; et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother Pharm. 2013, 72, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother Pharm. 2014, 73, 539–549. [Google Scholar] [CrossRef]
- Laquente, B.; Lopez-Martin, J.; Richards, D.; Illerhaus, G.; Chang, D.Z.; Kim, G.; Stella, P.; Richel, D.; Szcylik, C.; Cascinu, S.; et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 2017, 17, 137. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Takada, K.; Takeuchi, O.; Takagi, A.; Watanabe, K.; Hirohara, M.; Hamamoto, T.; Masuda, Y. Prexasertib increases the sensitivity of pancreatic cancer cells to gemcitabine and S-1. Oncol. Rep. 2020, 43, 689–699. [Google Scholar] [CrossRef]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rora, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Y.; Chen, S.; Kmieciak, M.; Leng, Y.; Lin, H.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A.; Dai, Y.; et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 2015, 29, 807–818. [Google Scholar] [CrossRef]
- Barbosa, R.S.S.; Dantonio, P.M.; Guimaraes, T.; de Oliveira, M.B.; Alves, V.L.F.; Sandes, A.F.; Fernando, R.C.; Colleoni, G.W.B. Sequential combination of bortezomib and WEE1 inhibitor, MK-1775, induced apoptosis in multiple myeloma cell lines. Biochem. Biophys. Res. Commun. 2019, 519, 597–604. [Google Scholar] [CrossRef]
- Ghelli Luserna Di Rora, A.; Beeharry, N.; Imbrogno, E.; Ferrari, A.; Robustelli, V.; Righi, S.; Sabattini, E.; Falzacappa, M.V.V.; Ronchini, C.; Testoni, N.; et al. Targeting WEE1 to enhance conventional therapies for acute lymphoblastic leukemia. J. Hematol. Oncol. 2018, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.R.W.; Langendonk, M.; Reitsma, B.; Herbers, P.; Nijland, M.; Huls, G.; van den Berg, A.; Ammatuna, E.; Visser, L.; van Meerten, T. WEE1 Inhibition Enhances Anti-Apoptotic Dependency as a Result of Premature Mitotic Entry and DNA Damage. Cancers 2019, 11, 1743. [Google Scholar] [CrossRef]
- Weisberg, E.; Nonami, A.; Chen, Z.; Liu, F.; Zhang, J.; Sattler, M.; Nelson, E.; Cowens, K.; Christie, A.L.; Mitsiades, C.; et al. Identification of Wee1 as a novel therapeutic target for mutant RAS-driven acute leukemia and other malignancies. Leukemia 2015, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.E.; Martin, M.M.; Feldman, D.S.; Terry, A.V., Jr.; Nuovo, G.J.; Elton, T.S. Experimental validation of miRNA targets. Methods 2008, 44, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Uchida, S.; Ishii, T.; Itaka, K.; Kataoka, K. Messenger RNA-based therapeutics for the treatment of apoptosis-associated diseases. Sci. Rep. 2015, 5, 15810. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Chang, P.; LeBlanc, A.; Li, D.; Abbruzzesse, J.L.; Frazier, M.L.; Killary, A.M.; Sen, S. MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev. Res. 2009, 2, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Du, Y.; Wang, G.; Gao, J.; Gong, Y.; Li, L.; Zhang, Z.; Zhu, J.; Jing, Q.; Qin, Y.; et al. Detection of differentially expressed microRNAs in serum of pancreatic ductal adenocarcinoma patients: miR-196a could be a potential marker for poor prognosis. Dig. Dis. Sci. 2011, 56, 602–609. [Google Scholar] [CrossRef]
- Szafranska, A.E.; Davison, T.S.; John, J.; Cannon, T.; Sipos, B.; Maghnouj, A.; Labourier, E.; Hahn, S.A. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene 2007, 26, 4442–4452. [Google Scholar] [CrossRef]
- Habbe, N.; Koorstra, J.B.; Mendell, J.T.; Offerhaus, G.J.; Ryu, J.K.; Feldmann, G.; Mullendore, M.E.; Goggins, M.G.; Hong, S.M.; Maitra, A. MicroRNA miR-155 is a biomarker of early pancreatic neoplasia. Cancer Biol. Ther. 2009, 8, 340–346. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sun, Y.W.; Liu, D.J.; Zhang, J.F.; Li, J.; Hua, R. MicroRNAs in stool samples as potential screening biomarkers for pancreatic ductal adenocarcinoma cancer. Am. J. Cancer Res. 2014, 4, 663–673. [Google Scholar] [PubMed]
- Kawaguchi, T.; Komatsu, S.; Ichikawa, D.; Morimura, R.; Tsujiura, M.; Konishi, H.; Takeshita, H.; Nagata, H.; Arita, T.; Hirajima, S.; et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br. J. Cancer 2013, 108, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, E.; Funel, N.; Peters, G.J.; del Chiaro, M.; Erozenci, L.A.; Vasile, E.; Leon, L.G.; Pollina, L.E.; Groen, A.; Falcone, A.; et al. MicroRNA-21 in pancreatic cancer: Correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010, 70, 4528–4538. [Google Scholar] [CrossRef]
- Li, Y.; VandenBoom, T.G., II; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Miura, S.; Satoh, K.; Shimosegawa, T. MiR-365 induces gemcitabine resistance in pancreatic cancer cells by targeting the adaptor protein SHC1 and pro-apoptotic regulator BAX. Cell Signal. 2014, 26, 179–185. [Google Scholar] [CrossRef]
- Wong, K.K. DNMT1 as a therapeutic target in pancreatic cancer: Mechanisms and clinical implications. Cell Oncol. 2020, 43, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, S.; Alcala, S.; Bayon, G.F.; Kheir, T.B.; Schoenhals, M.; González-Neira, A.; Fraga, M.F.; Aicher, A.; Heeschen, C.; Sainz, B., Jr. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17-92 Cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [PubMed]
- Rosty, C.; Geradts, J.; Sato, N.; Wilentz, R.E.; Roberts, H.; Sohn, T.; Cameron, J.L.; Yeo, C.J.; Hruban, R.H.; Goggins, M. Inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am. J. Surg. Pathol. 2003, 27, 1495–1501. [Google Scholar] [CrossRef]
- Sato, N.; Fukushima, N.; Maitra, A.; Matsubayashi, H.; Yeo, C.J.; Cameron, J.L.; Hruban, R.H.; Goggins, M. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003, 63, 3735–3742. [Google Scholar] [PubMed]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharm. 2014, 74, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, S.; Liu, H.M.; Zhang, Y.B.; Blair, C.A.; Mercola, D.; Sassone-Corsi, P.; Zi, X. Histone lysine-specific methyltransferases and demethylases in carcinogenesis: New targets for cancer therapy and prevention. Curr. Cancer Drug Targets 2013, 13, 558–579. [Google Scholar] [CrossRef]
- Richards, D.A.; Boehm, K.A.; Waterhouse, D.M.; Wagener, D.J.; Krishnamurthi, S.S.; Rosemurgy, A.; Grove, W.; Macdonald, K.; Gulyas, S.; Clark, M.; et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: Results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann. Oncol. 2006, 17, 1096–1102. [Google Scholar] [CrossRef]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, Q.; Dudek, A.Z. Phase II study of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy. Anticancer Res. 2012, 32, 1027–1031. [Google Scholar] [PubMed]
- Wang, Y.H.; Sui, Y.N.; Yan, K.; Wang, L.S.; Wang, F.; Zhou, J.H. BRD4 promotes pancreatic ductal adenocarcinoma cell proliferation and enhances gemcitabine resistance. Oncol. Rep. 2015, 33, 1699–1706. [Google Scholar] [CrossRef]
- Sahai, V.; Kumar, K.; Knab, L.; Chow, C.; Raza, S.; Bentrem, D.; Ebine, K.; Munshi, H. BET Bromodomain Inhibitors Block Growth of Pancreatic Cancer Cells in Three-Dimensional Collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Huang, M.; Lin, X.; Liu, C.; Liu, Z.; Meng, F.; Wang, C.; Huang, Q. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci. Rep. 2018, 8, 8102. [Google Scholar] [CrossRef]
- Falchook, G.; Rosen, S.; LoRusso, P.; Watts, J.; Gupta, S.; Coombs, C.C.; Talpaz, M.; Kurzrock, R.; Mita, M.; Cassaday, R.; et al. Development of 2 Bromodomain and Extraterminal Inhibitors with Distinct Pharmacokinetic and Pharmacodynamic Profiles for the Treatment of Advanced Malignancies. Clin. Cancer Res. 2020, 26, 1247–1257. [Google Scholar] [CrossRef]
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307. [Google Scholar]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Brana, I.; Cassier, P.A.; Moreno, V.; de Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr. 2020, 4, pkz093. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial with Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF—MEK—ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.t.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Ruggeri, B.A.; Huang, L.; Wood, M.; Cheng, J.Q.; Testa, J.R. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 1998, 21, 81–86. [Google Scholar] [CrossRef]
- Konstantinidou, G.; Bey, E.A.; Rabellino, A.; Schuster, K.; Maira, M.S.; Gazdar, A.F.; Amici, A.; Boothman, D.A.; Scaglioni, P.P. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009, 69, 7644–7652. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Aung, W.; Tsuji, A.B.; Sudo, H.; Sugyo, A.; Ukai, Y.; Kouda, K.; Kurosawa, Y.; Furukawa, T.; Saga, T.; Higashi, T. Combined treatment of pancreatic cancer xenograft with (90)Y-ITGA6B4-mediated radioimmunotherapy and PI3K/mTOR inhibitor. World J. Gastroenterol. 2017, 23, 7551–7562. [Google Scholar] [CrossRef]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef]
- McRee, A.J.; Sanoff, H.K.; Carlson, C.; Ivanova, A.; O’Neil, B.H. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Investig. New Drugs 2015, 33, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; van Cutsem, E.; Perez-Garcia, J.; Stathis, A.; Britten, C.D.; et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; LoRusso, P.; Kwak, E.; Pandya, S.; Rudin, C.M.; Kurkjian, C.; Cleary, J.M.; Pilat, M.J.; Jones, S.; de Crespigny, A.; et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 419–432. [Google Scholar] [CrossRef]
- Gaudio, E.; Tarantelli, C.; Kwee, I.; Barassi, C.; Bernasconi, E.; Rinaldi, A.; Ponzoni, M.; Cascione, L.; Targa, A.; Stathis, A.; et al. Combination of the MEK inhibitor pimasertib with BTK or PI3K-delta inhibitors is active in preclinical models of aggressive lymphomas. Ann. Oncol. 2016, 27, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Dean, E.; Puglisi, M.; Greystoke, A.; Ong, M.; Burke, W.; Cavallin, M.; Bigley, G.; Womack, C.; Harrington, E.A.; et al. First-in-Human Pharmacokinetic and Pharmacodynamic Study of the Dual m-TORC 1/2 Inhibitor AZD2014. Clin. Cancer Res. 2015, 21, 3412–3419. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; O’Reilly, E.M.; Brody, J.R.; Witkiewicz, A.K. Genetic Diversity of Pancreatic Ductal Adenocarcinoma and Opportunities for Precision Medicine. Gastroenterology 2016, 150, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, T.; Liao, Q.; Dai, M.; Guo, J.; Yang, X.; Tan, W.; Lin, D.; Wu, C.; Zhao, Y. Metformin inhibits pancreatic cancer metastasis caused by SMAD4 deficiency and consequent HNF4G upregulation. Protein Cell 2021, 12, 128–144. [Google Scholar] [CrossRef]
- Zwick, E.; Bange, J.; Ullrich, A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr. Relat. Cancer 2001, 8, 161–173. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Ségaliny, A.I.; Tellez-Gabriel, M.; Heymann, M.F.; Heymann, D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J. Bone Oncol. 2015, 4, 1–12. [Google Scholar] [CrossRef]
- Pettazzoni, P.; Viale, A.; Shah, P.; Carugo, A.; Ying, H.; Wang, H.; Genovese, G.; Seth, S.; Minelli, R.; Green, T.; et al. Genetic events that limit the efficacy of MEK and RTK inhibitor therapies in a mouse model of KRAS-driven pancreatic cancer. Cancer Res. 2015, 75, 1091–1101. [Google Scholar] [CrossRef]
- Miyabayashi, K.; Ijichi, H.; Mohri, D.; Tada, M.; Yamamoto, K.; Asaoka, Y.; Ikenoue, T.; Tateishi, K.; Nakai, Y.; Isayama, H.; et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res. 2013, 73, 2221–2234. [Google Scholar] [CrossRef]
- Schultheis, B.; Reuter, D.; Ebert, M.P.; Siveke, J.; Kerkhoff, A.; Berdel, W.E.; Hofheinz, R.; Behringer, D.M.; Schmidt, W.E.; Goker, E.; et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen in KRAS wildtype patients with locally advanced or metastatic pancreatic cancer: A multicenter, randomized phase IIb study. Ann. Oncol. 2017, 28, 2429–2435. [Google Scholar] [CrossRef]
- Skrypek, N.; Vasseur, R.; Vincent, A.; Duchene, B.; van Seuningen, I.; Jonckheere, N. The oncogenic receptor ErbB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget 2015, 6, 10853–10867. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Konstantinidou, G.; Ramadori, G.; Torti, F.; Kangasniemi, K.; Ramirez, R.E.; Cai, Y.; Behrens, C.; Dellinger, M.T.; Brekken, R.A.; Wistuba, I.I.; et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013, 3, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol Ther. 2018, 19, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Liu, X.; Knolhoff, B.L.; Hegde, S.; Lee, K.B.; Jiang, H.; Fields, R.C.; Pachter, J.A.; Lim, K.H.; De Nardo, D.G. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 2020, 69, 122–132. [Google Scholar] [CrossRef]
- Turner, H.; Kinet, J.P. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature 1999, 402 (Suppl. S6760), B24–B30. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.; Javle, M.; Davis, R.E.; Vats, P.; Kumar-Sinha, C.; Xiao, L.; Mettu, N.B.; Parra, E.R.; Benson, A.B.; Lopez, C.D.; et al. Randomized phase II study of the Bruton tyrosine kinase inhibitor acalabrutinib, alone or with pembrolizumab in patients with advanced pancreatic cancer. J. Immunother. Cancer 2020, 8, e000587. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Vervenne, W.L.; Bennouna, J.; Humblet, Y.; Gill, S.; van Laethem, J.L.; Verslype, C.; Scheithauer, W.; Shang, A.; Cosaert, J.; et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2009, 27, 2231–2237. [Google Scholar] [CrossRef]
- Kindler, H.L.; Ioka, T.; Richel, D.J.; Bennouna, J.; Letourneau, R.; Okusaka, T.; Funakoshi, A.; Furuse, J.; Park, Y.S.; Ohkawa, S.; et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: A double-blind randomised phase 3 study. Lancet Oncol. 2011, 12, 256–262. [Google Scholar] [CrossRef]
- Goncalves, A.; Gilabert, M.; Francois, E.; Dahan, L.; Perrier, H.; Lamy, R.; Re, D.; Largillier, R.; Gasmi, M.; Tchiknavorian, X.; et al. BAYPAN study: A double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann. Oncol. 2012, 23, 2799–2805. [Google Scholar] [CrossRef]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; de Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Pozzato, C.; Saliakoura, M.; Yang, Z.; Peng, R.W.; Galie, M.; Oberson, K.; Simon, H.U.; Karamitopoulou, E.; Konstantinidou, G. ACSL3-PAI-1 signaling axis mediates tumor-stroma cross-talk promoting pancreatic cancer progression. Sci. Adv. 2020, 6, eabb9200. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Vaupel, P.; Thews, O.; Kelleher, D.K. Pancreatic tumors show high levels of hypoxia: Regarding Koong et al. IJROBP 2000;48:919-922. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 1099–1100. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, K.Y.; Rivera, L.B.; Hur, H.; Carbon, J.G.; Toombs, J.E.; Goldstein, C.D.; Dellinger, M.T.; Castrillon, D.H.; Brekken, R.A. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014, 74, 1032–1044. [Google Scholar] [CrossRef]
- Michalopoulou, E.; Bulusu, V.; Kamphorst, J.J. Metabolic scavenging by cancer cells: When the going gets tough, the tough keep eating. Br. J. Cancer 2016, 115, 635–640. [Google Scholar] [CrossRef]
- Saliakoura, M.; Sebastiano, M.R.; Pozzato, C.; Heidel, F.H.; Schnoder, T.M.; Prince, S.S.; Bubendorf, L.; Pinton, P.; Schmid, R.A.; Baumgartner, J.; et al. PLCgamma1 suppression promotes the adaptation of KRAS-mutant lung adenocarcinomas to hypoxia. Nat. Cell Biol. 2020, 22, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Heiden, M.G.V.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef]
- Saliakoura, M.; Reynoso-Moreno, I.; Pozzato, C.; Sebastiano, M.R.; Galie, M.; Gertsch, J.; Konstantinidou, G. The ACSL3-LPIAT1 signaling drives prostaglandin synthesis in non-small cell lung cancer. Oncogene 2020, 39, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Lima, C.M.R.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharm. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- McNeillis, R.; Greystoke, A.; Walton, J.; Bacon, C.; Keun, H.; Siskos, A.; Petrides, G.; Leech, N.; Jenkinson, F.; Bowron, A.; et al. A case of malignant hyperlactaemic acidosis appearing upon treatment with the mono-carboxylase transporter 1 inhibitor AZD3965. Br. J. Cancer 2020, 122, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.; Ranson, M.; Kelso, M.J. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: An orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer 2011, 129, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Jayashankar, V.; Edinger, A.L. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat. Commun. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef]
- Tschan, M.P.; Simon, H.U. The role of autophagy in anticancer therapy: Promises and uncertainties. J. Intern. Med. 2010, 268, 410–418. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Haist, M.; Stege, H.; Grabbe, S.; Bros, M. The Functional Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells within the Immunosuppressive Tumor Microenvironment. Cancers 2021, 13, 210. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.M.; Michl, P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2017, 66, 1278–1285. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed]
- Plate, J.M.; Plate, A.E.; Shott, S.; Bograd, S.; Harris, J.E. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol. Immunother. 2005, 54, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J., III; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Soreide, O.; Eriksen, J.A.; Moller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Weden, S.; Klemp, M.; Gladhaug, I.P.; Moller, M.; Eriksen, J.A.; Gaudernack, G.; Buanes, T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int. J. Cancer 2011, 128, 1120–1128. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Shima, H.; Tsurita, G.; Wada, S.; Hirohashi, Y.; Yasui, H.; Hayashi, H.; Miyakoshi, T.; Watanabe, K.; Murai, A.; Asanuma, H.; et al. Randomized phase II trial of survivin 2B peptide vaccination for patients with HLA-A24-positive pancreatic adenocarcinoma. Cancer Sci. 2019, 110, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Mori, M.; Yoshizaki, S.; Tsukinaga, S.; Odahara, S.; Koyama, S.; Imazu, H.; et al. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin. Cancer Res. 2014, 20, 4228–4239. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A., III; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef] [PubMed]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagala, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef]
- Goldman, J.W.; Waterhouse, D.M.; George, B.; O’Dwyer, P.J.; Bhore, R.; Banerjee, S.; Lyons, L.; Louis, C.U.; Ong, T.J.; Kelly, K. Safety and Efficacy Results of a Phase I, Open-Label Study of Concurrent and Delayed Nivolumab in Combination With nab-Paclitaxel and Carboplatin in Advanced Non-small Cell Lung Cancer. Front. Oncol. 2019, 9, 1256. [Google Scholar] [CrossRef]
- Chung, M.J.; Park, J.Y.; Bang, S.; Park, S.W.; Song, S.Y. Phase II clinical trial of ex vivo-expanded cytokine-induced killer cells therapy in advanced pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Lum, L.G.; Thakur, A.; Choi, M.; Deol, A.; Kondadasula, V.; Schalk, D.; Fields, K.; Dufrense, M.; Philip, P.; Dyson, G.; et al. Clinical and immune responses to anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (EGFR BATs) in pancreatic cancer patients. Oncoimmunology 2020, 9, 1773201. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chapman, P.B.; Feilchenfeldt, J.; Brennan, M.F.; Capanu, M.; Gansukh, B.; Jacobs, G.; Levin, A.; Neville, D.; Kelsen, D.P.; et al. Targeting mutated K-ras in pancreatic adenocarcinoma using an adjuvant vaccine. Am. J. Clin. Oncol. 2011, 34, 321–325. [Google Scholar] [CrossRef]
- Kubo, T.; Tsurita, G.; Hirohashi, Y.; Yasui, H.; Ota, Y.; Watanabe, K.; Murai, A.; Matsuo, K.; Asanuma, H.; Shima, H.; et al. Immunohistological analysis of pancreatic carcinoma after vaccination with survivin 2B peptide: Analysis of an autopsy series. Cancer Sci. 2019, 110, 2386–2395. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.H.; Diamond, M.S.; Hay, C.A.; Byrne, K.T.; Vonderheide, R.H. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 8022–8031. [Google Scholar] [CrossRef]
- Tahkola, K.; Ahtiainen, M.; Kellokumpu, I.; Mecklin, J.P.; Laukkarinen, J.; Laakkonen, J.; Kenessey, I.; Jalkanen, S.; Salmi, M.; Bohm, J. Prognostic impact of CD73 expression and its relationship to PD-L1 in patients with radically treated pancreatic cancer. Virchows Arch. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharm. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef]
- Cheent, K.; Khakoo, I.S. Natural killer cells: Integrating diversity with function. Immunology 2009, 126, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, G.; Rimessi, A. Editorial: Oncogenic RAS-Dependent Reprogramming of Cellular Plasticity. Front. Oncol. 2020, 10, 588. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Targeted Pathway | Phase/Patient Number | Intervention | Subject Population | Results | Refs |
|---|---|---|---|---|---|---|
| PEGPH20 | HA | Phase 1b/II n = 138 | mFOLFIRINOX | Metastatic PCa | Grade 3 to 4 toxicity and worsened OS | NCT01959139 [127] |
| PEGPH20 | HA | Phase III n = 494 | Gem + nab-PTX | Metastatic PCa | Grade 3 to 4 toxicity and worsened OS | NCT02715804 [128] |
| Vismodegib | Hedgehog pathway | Phase Ib/II n = 106 | Gem | Metastatic PCa | Does not improve overall response rate, PFS, or OS | NCT01064622 [129] |
| Vismodegib | Hedgehog pathway | Phase II n = 71 | Gem + nab-PTX | Metastatic PCa | Does not improve overall response rate, PFS, or OS | NCT01088815 [130] |
| Bevacizumab | VEGF pathway | Phase III n = 535 | Gem | Advanced PCa | Does not improve OS | NCT00088894 [131] |
| Bevacizumab | VEGF pathway | Phase III n = 607 | Gem + erlotinib | Metastatic PCa | Does not improve OS, improved PFS | NCT01214720 [132] |
| Axitinib | VEGF pathway | Phase III n = 632 | Gem | Advanced PCa | Does not improve OS, improved PFS | NCT00471146 [133] |
| Sorefenib | VEGF, PDGF and RAF pathway | Phase III n = 102 | Gem | Advanced PCa | Does not improve overall response rate, PFS, or OS | NCT00541021 [134] |
| HCQ | Autophagy | Phase II/n = 98 | Gem + nab-PTX | Preoperative PCa | Greater tumor response, improved serum biomarker response, and immune activity | NCT01978184 [135] |
| HCQ | Autophagy | Phase II/n = 112 | Gem + nab-PTX | Metastatic PCa | Greater pathological tumor response, but not OS | NCT01506973 [136] |
| Drug | Targeted Pathway | Phase/Patient # | Treatment Combo | PCa | Result | Refs |
|---|---|---|---|---|---|---|
| Cancer vaccines | ||||||
| GVAX | T cell response | Phase IIb n = 169 | CP + CRS-207 | Previously treated metastatic | Well tolerated, does not improve OS vs chemotherapy | NCT02004262 [164] |
| Peptide vaccine | mRAS | Phase I/II n = 38 | GM-CSF as adjuvant | Advanced | Durature memory against mRAS, activation of RAS-specific T cells, improved OS vs non responders | CTN-97004 [165] |
| Peptide vaccine | mRAS | 10 year follow-up n = 11 | GM-CSF as adjuvant | Advanced/lymph node metastases | Durature memory against mRAS (years) positive immune response in all patients | CTN-98010 [166] |
| Peptide vaccine (GI-4000) | hTERT | Phase III n = 1062 | Gem+ Cap | Advanced/Metastatic | Well tolerated, does not improve OS | [167] |
| Peptide vaccine (SVN-2B) | HLA-A24 of survivin 2B | Phase II n = 83 | IFNβ | Advanced | SVN-2B + IFNβ improved OS and immunological reaction vs placebo | UMIN000012146 [168] |
| DC-based vaccine | WT1 | Phase I n = 10 | Gem | Advanced | Increased OS, PFS and specific T cell responses. | UMIN00004063 [169] |
| Monoclonal antibodies | ||||||
| CD40 monoclonal antibody (CP-870,893) | T cell responses | Phase I n= 21 | Gem | CT-naïve, unresectable/metastatic | Mild cytokine release syndrome, 4/21 PR, 11/21 SD, and 4/21 PD, improved OS and PFS vs Gem | NCT00711191 [170] |
| Immune checkpoint inhibitors | ||||||
| Ipilimumab | CTLA-4 | Phase Ib n = 16 | Gem | Advanced | Grade 3 to 4 hematologic adverse events, 2/16 PR, 5/16 SD, does not improve OS | NCT01473940 [171] |
| Tremelimumab (CP-675,206) | CTLA-4 | Phase I n = 34 | Gem | Advanced | Grade 3 to 4 hematologic adverse events, improved OS vs historical Gem results | NCT00556023 [172] |
| Ipilimumab | CTLA-4 | Phase I n = 30 | GVAX | Previously treated, advanced | 3/17 SD, 7/15 reduced CA19-9, improved OS vs ipilimumab alone | NCT00836407 [173] |
| Pembrolizumab | PD-1 | Phase Ib/II n = 17 (11 evaluable) | Gem + nab-PTX | Metastatic | Grade 3 to 4 hematologic adverse events, 3/11 PR, 8 SD | NCT02331251 [174] |
| Nivolumab/nab-PTX | PD-1 | Phase I n= 44 | Gem | Advanced | 2% CR, 16% PR, 46% SD ≥ 6 weeks, 20% PD | NCT02309177 [175] |
| Adoptive cell therapy/CAR-T | ||||||
| Cytokine-induced killer cells | MHC- antitumor activity | Phase II/n = 20 | Second-line Gem refractory advanced | Does not improve OS or PFS, but improves quality of life | NCT00965718 [176] | |
| CAR-T | Mesothelin | Phase I/n = 6 | CT-refractory metastatic | SD (2/6) | NCT01897415 [177] | |
| CAR-T | Mesothelin | Phase I/II n = 7 | anti-CD3 + anti-EGFR | Advanced/Metastatic | Anti-cancer cytotoxicity and increased innate immune responses | NCT02620865 [178] |
| Targeted Mechanism | Targeted Pathway |
|---|---|
| DNA Repairing Deficiency and Microsatellite Instability | PARP |
| ATM | |
| ATR | |
| DNA-PK | |
| CHK1/2 | |
| Wee1 | |
| Epigenetic Alterations | miRNAs |
| DNA methyltransferase 1 | |
| HATs and HDACs | |
| Bromodomain proteins | |
| Key Signaling Pathways | KRAS, PI3K, mTOR |
| TP53 | |
| SMAD4 | |
| Tyrosine Kinase Signaling (EGFR, HER2, FAK, BTK) | |
| Tumor Microenvironment and Related Metabolic Reprogramming | HA |
| Hedgehog | |
| VEGF | |
| Glycolysis, monocarboxylate transporters | |
| Autophagy | |
| Immune Regulatory Networks | GM-CSF |
| Mutated KRAS peptides (T cell response) | |
| hTERT peptides (T cell response) | |
| Survivin peptides (T cell response) | |
| DCs (WT1) | |
| CD40 | |
| CTLA-4 | |
| PD-1/PD-L1 | |
| CD73 | |
| CD3/EGFR | |
| PSCA (ongoing) | |
| CEA+ (ongoing) | |
| CD133 (ongoing) | |
| Nectin 4/FAP (ongoing) | |
| CSF1/CSF1R (ongoing) | |
| CXCR-4 (ongoing) | |
| Pluripotent stem cell-derived NK cells (ongoing) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leroux, C.; Konstantinidou, G. Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology. Cancers 2021, 13, 799. https://doi.org/10.3390/cancers13040799
Leroux C, Konstantinidou G. Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology. Cancers. 2021; 13(4):799. https://doi.org/10.3390/cancers13040799
Chicago/Turabian StyleLeroux, Cédric, and Georgia Konstantinidou. 2021. "Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology" Cancers 13, no. 4: 799. https://doi.org/10.3390/cancers13040799
APA StyleLeroux, C., & Konstantinidou, G. (2021). Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology. Cancers, 13(4), 799. https://doi.org/10.3390/cancers13040799