
Cache Domain Containing 1 Is a Novel Marker of Non-Alcoholic Steatohepatitis-Associated Hepatocarcinogenesis

 ,
,  , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
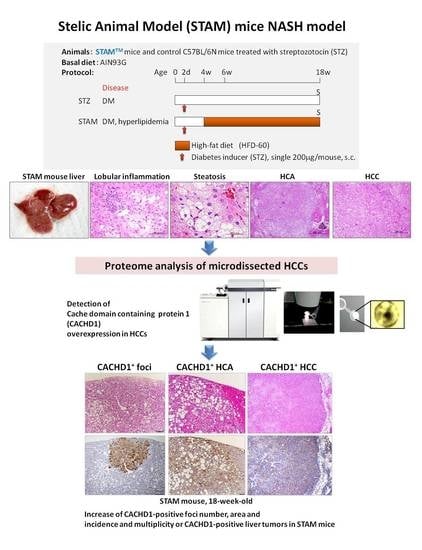
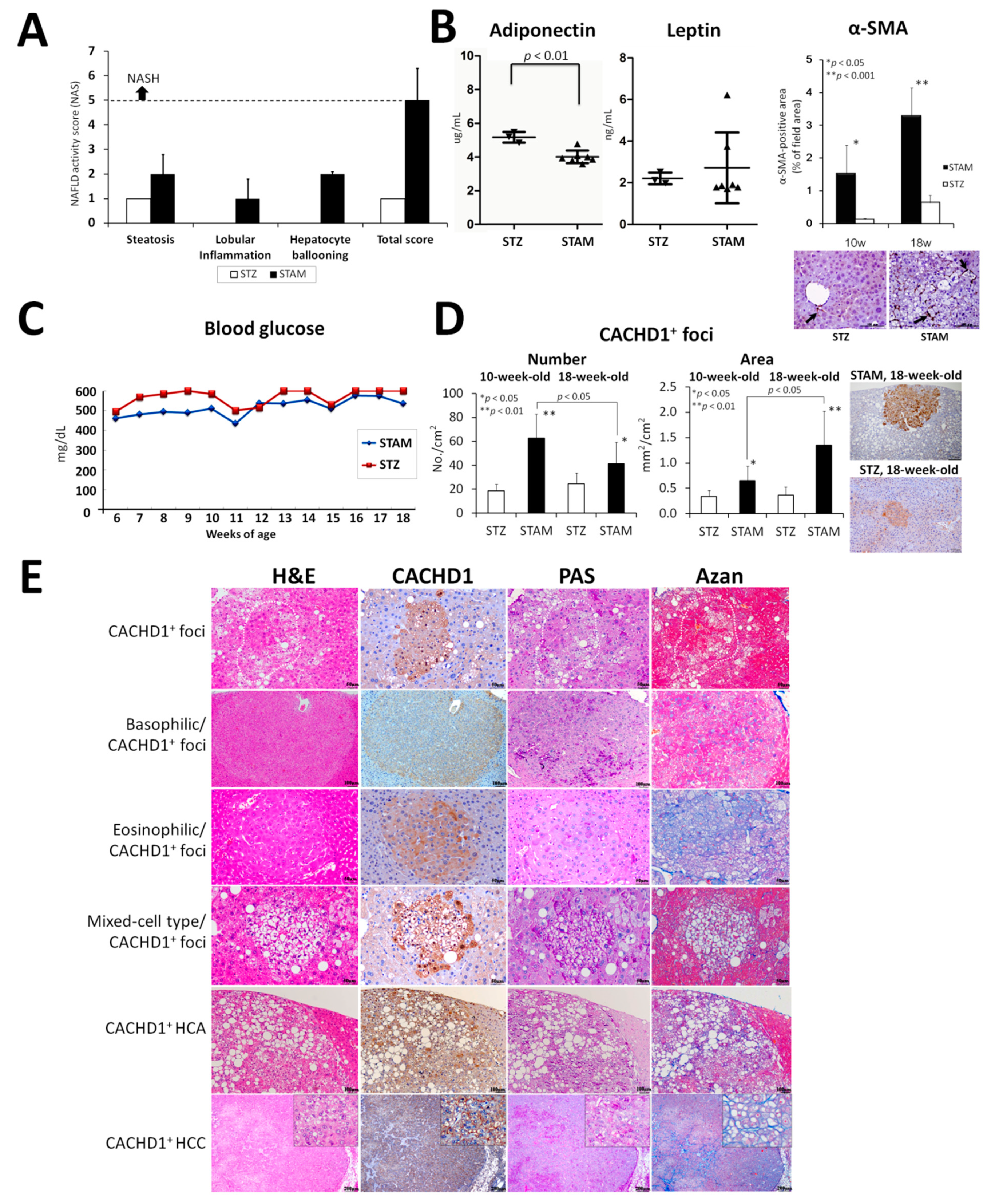
2.1. STAM Mice NASH Model
2.1.1. General Conditions
2.1.2. Histopathological Analysis
2.1.3. Proteome Analysis of STAM Mice HCCs
2.1.4. Immunohistochemical Assessment of CACHD1 in Mice Livers
2.2. In Vitro Functional Analysis
2.2.1. Effects Induced by CACHD1siRNAs Knockdown in Huh7 and HepG2 Cells
2.2.2. Proteome and Ingenuity Pathway Analyses of CACHD1 Knockdown Huh7 and HepG2 Cell Lines
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. STAM Mice Experiment
4.3. Proteome Analysis in STAM Mice HCCs
4.4. Immunohistochemical Examination
4.5. In Vitro Experiments
4.5.1. Cell Lines and Culture Conditions
4.5.2. CACHD1 siRNA Knockdown in Huh7 and HepG2 Human Liver Cancer Cells
4.5.3. Real-Time Quantitative PCR
4.5.4. Protein Extraction and Western Blot Analysis
4.5.5. WST-8 Assay
4.5.6. QSTAR LC-Ms/Ms and Ingenuity Pathway Analysis (IPA)
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Kakehashi, A.; Stefanov, V.E.; Ishii, N.; Okuno, T.; Fujii, H.; Kawai, K.; Kawada, N.; Wanibuchi, H. Proteome characteristics of non-alcoholic steatohepatitis liver tissue and associated hepatocellular carcinomas. Int. J. Mol. Sci. 2017, 18, 434. [Google Scholar] [CrossRef]
- Hill-Baskin, A.E.; Markiewski, M.M.; Buchner, D.A.; Shao, H.; DeSantis, D.; Hsiao, G.; Subramaniam, S.; Berger, N.A.; Croniger, C.M.; Lambris, J.D.; et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum. Mol. Genet. 2009, 18, 2975–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Bando, I.; Mennear, J.H.; Bernard, B.K. Studies of the toxicological potential of tripeptides (L-Valyl-L-prolyl-L-proline and L-lsoleucyl-L-prolyl-L-proline): IV. Assessment of the repeated-dose toxicological potential of synthesized L-Valyl-L-prolyl-L-proline in male and female rats and dogs. Int. J. Toxicol. 2005, 24 (Suppl. 4), 25–39. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Tsuneyama, K.; Fujimoto, M.; Nomoto, K.; Hayashi, S.; Miwa, S.; Nakajima, T.; Nakanishi, Y.; Sasaki, Y.; Suzuki, W.; et al. Spontaneous onset of nonalcoholic steatohepatitis and hepatocellular carcinoma in a mouse model of metabolic syndrome. Lab. Investig. 2013, 93, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Tabuchi, M.; Suzuki, W.; Iizuka, S.; Nagata, M.; Ikeya, Y.; Takeda, S.; Shimada, T.; Aburada, M. Insulin resistance and low sympathetic nerve activity in the Tsumura Suzuki obese diabetic mouse: A new model of spontaneous type 2 diabetes mellitus and obesity. Metabolism 2006, 55, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Kakehashi, A.; Ishii, N.; Fujioka, M.; Gi, M.; Wanibuchi, H. mTOR activation in liver tumors is associated with metabolic syndrome and non-alcoholic steatohepatitis in both mouse models and humans. Cancers 2018, 10, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptozotocin. Mutat. Res. 2002, 512, 121–134. [Google Scholar] [CrossRef]
- Sugiura, M.; Ohshima, M.; Ogawa, K.; Yano, M. Chronic administration of Satsuma Mandarin fruit (Citrus unshiu MARC.) improves oxidative stress in streptozotocin-induced diabetic rat liver. Biol. Pharm. Bull. 2006, 29, 588–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Muramatsu, M.; Ishii, Y.; Saigo, Y.; Konuma, T.; Toriniwa, Y.; Miyajima, K.; Ohta, T. Pathophysiological analysis of the progression of hepatic lesions in STAM mice. Physiol. Res. 2017, 66, 791–799. [Google Scholar] [CrossRef]
- Dahimene, S.; Page, K.M.; Kadurin, I.; Ferron, L.; Ho, D.Y.; Powell, G.T.; Pratt, W.S.; Wilson, S.W.; Dolphin, A.C. The alpha2delta-like protein Cachd1 increases N-type calcium currents and cell surface expression and competes with alpha2delta. Cell Rep. 2018, 25, 1610–1621.e5. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, G.S.; Soubrane, C.H.; Hounshell, J.A.; Lin, H.; Owenson, V.; Rigby, M.; Cox, P.J.; Barker, B.S.; Ottolini, M.; Ince, S.; et al. CACHD1 is an alpha2delta-like protein that modulates CaV3 voltage-gated calcium channel activity. J. Neurosci. 2018, 38, 9186–9201. [Google Scholar] [CrossRef] [Green Version]
- Stephens, G.J.; Cottrell, G.S. CACHD1: A new activity-modifying protein for voltage-gated calcium channels. Channels 2019, 13, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakehashi, A. (Dept. Mol. Pathology, Osaka City University Graduate School of Medicine, Osaka, Japan). IHC in STAM and control C57Bl/6N mice liver. Unpublished work. 2021. [Google Scholar]
- Masouminia, M.; Samadzadeh, S.; Ebaee, A.; French, B.; Tillman, B.; French, S. Alcoholic steatohepatitis (ASH) causes more UPR-ER stress than non-alcoholic steatohepatitis (NASH). Exp. Mol. Pathol. 2016, 101, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njavro, J.R.; Klotz, J.; Dislich, B.; Wanngren, J.; Shmueli, M.D.; Herber, J.; Kuhn, P.; Kumar, R.; Koeglsperger, T.; Conrad, M.; et al. Mouse brain proteomics establishes MDGA1 and CACHD1 as in vivo substrates of the Alzheimer protease BACE1. FASEB J. 2020, 34, 2465–2482. [Google Scholar] [CrossRef]
- Guner, G.; Lichtenthaler, S.F. The substrate repertoire of gamma-secretase/presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Dragin, N.; Shi, Z.; Madan, R.; Karp, C.L.; Sartor, M.A.; Chen, C.; Gonzalez, F.J.; Nebert, D.W. Phenotype of the Cyp1a1/1a2/1b1-/- triple-knockout mouse. Mol. Pharmacol. 2008, 73, 1844–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016, 17, 1206. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, J.Z.; Chung, J.; Purdom, E.; Wang, N.J.; Kakavand, H.; Wilmott, J.S.; Butler, T.; Thompson, J.F.; Mann, G.J.; Haydu, L.E.; et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. USA 2015, 112, 10995–11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef]
- Tolomeo, D.; Agostini, A.; Macchia, G.; L’Abbate, A.; Severgnini, M.; Cifola, I.; Frassanito, M.A.; Racanelli, V.; Solimando, A.G.; Haglund, F.; et al. BL1391: An established cell line from a human malignant peripheral nerve sheath tumor with unique genomic features. Hum. Cell 2021, 34, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Zhu, H.H.; Zhu, Y.M. Diabetes and cancer: Associations, mechanisms, and implications for medical practice. World J. Diabetes 2014, 5, 372–380. [Google Scholar] [CrossRef]
- Yoshimine, Y.; Uto, H.; Kumagai, K.; Mawatari, S.; Arima, S.; Ibusuki, R.; Mera, K.; Nosaki, T.; Kanmura, S.; Numata, M.; et al. Hepatic expression of the Sptlc3 subunit of serine palmitoyltransferase is associated with the development of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis. Oncol. Rep. 2015, 33, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiburek, L.; Cesnekova, J.; Kostkova, O.; Fornuskova, D.; Vinsova, K.; Wenchich, L.; Houstek, J.; Zeman, J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 2012, 23, 1010–1023. [Google Scholar] [CrossRef]
- Kinoshita, A.; Wanibuchi, H.; Imaoka, S.; Ogawa, M.; Masuda, C.; Morimura, K.; Funae, Y.; Fukushima, S. Formation of 8-hydroxydeoxyguanosine and cell-cycle arrest in the rat liver via generation of oxidative stress by phenobarbital: Association with expression profiles of p21(WAF1/Cip1), cyclin D1 and Ogg1. Carcinogenesis 2002, 23, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, X.; Tan, H.-Y.; Li, S.; Tsang, C.M.; Tsao, S.-W.; Feng, Y. Berberine suppresses cyclin D1 expression through proteasomal degradation in human hepatoma cells. Int. J. Mol. Sci. 2016, 17, 1899. [Google Scholar] [CrossRef] [Green Version]
- Iida, A.; Kuranuki, S.; Yamamoto, R.; Uchida, M.; Ohta, M.; Ichimura, M.; Tsuneyama, K.; Masaki, T.; Seike, M.; Nakamura, T. Analysis of amino acid profiles of blood over time and biomarkers associated with non-alcoholic steatohepatitis in STAM mice. Exp. Anim. 2019, 68, 417–428. [Google Scholar] [CrossRef]
- Orime, K.; Shirakawa, J.; Togashi, Y.; Tajima, K.; Inoue, H.; Nagashima, Y.; Terauchi, Y. Lipid-lowering agents inhibit hepatic steatosis in a non-alcoholic steatohepatitis-derived hepatocellular carcinoma mouse model. Eur. J. Pharmacol. 2016, 772, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Ferguson, D.A.; Lee, C.I.; Proksch, J.W. Omaveloxolone and TX63682 are hepatoprotective in the STAM mouse model of nonalcoholic steatohepatitis. J. Biochem. Mol. Toxicol. 2020, 34, e22526. [Google Scholar] [CrossRef]
- Liebig, M.; Dannenberger, D.; Vollmar, B.; Abshagen, K. n-3 PUFAs reduce tumor load and improve survival in a NASH-tumor mouse model. Ther. Adv. Chronic Dis. 2019, 10, 2040622319872118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandla, H.; Dasgupta, D.; Mauer, A.S.; Nozickova, B.; Kumar, S.; Hirsova, P.; Graham, R.P.; Malhi, H. Deletion of endoplasmic reticulum stress-responsive co-chaperone p58(IPK) protects mice from diet-induced steatohepatitis. Hepatol. Res. 2018, 48, 479–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.L.; Shajahan, A.N.; Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer Ther. 2011, 11, 1283–1294. [Google Scholar] [CrossRef]
- Warri, A.; Cook, K.L.; Hu, R.; Jin, L.; Zwart, A.; Soto-Pantoja, D.R.; Liu, J.; Finkel, T.; Clarke, R. Autophagy and unfolded protein response (UPR) regulate mammary gland involution by restraining apoptosis-driven irreversible changes. Cell Death Discov. 2018, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Kakehashi, A.; Ishii, N.; Shibata, T.; Wei, M.; Okazaki, E.; Tachibana, T.; Fukushima, S.; Wanibuchi, H. Mitochondrial prohibitions and septin 9 are implicated in the onset of rat hepatocarcinogenesis. Toxicol. Sci. 2010, 119, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakehashi, A.; Hagiwara, A.; Imai, N.; Nagano, K.; Nishimaki, F.; Banton, M.; Wei, M.; Fukushima, S.; Wanibuchi, H. Mode of action of ethyl tertiary-butyl ether hepatotumorigenicity in the rat: Evidence for a role of oxidative stress via activation of CAR, PXR and PPAR signaling pathways. Toxicol Appl Pharmacol. 2013, 273, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Wei, M.; Yamano, S.; Kakehashi, A.; Tamada, S.; Nakatani, T.; Wanibuchi, H. DDX39 acts as a suppressor of invasion for bladder cancer. Cancer Sci. 2012, 103, 1363–1369. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group/Duration | No. Mice | Incidence (%) | Multiplicity (No./Mouse) | ||||
|---|---|---|---|---|---|---|---|
| HCA | HCC | Total | HCA | HCC | Total | ||
| STZ | 4 | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 |
| STAM | 7 | 7 (100) ** | 5 (71) * | 7 (100) ** | 2.7 ± 1.7 * | 1.0 ± 0.8 * | 3.7 ± 2.1 * |
| Name | GI Number | Ratio | p Value | Location | Functions |
|---|---|---|---|---|---|
| Cache domain-containing protein 1 (CACHD1) | 39930563 | 3.63 | <0.001 | C, M | Ca T |
| Cytokeratin, type II cytoskeletal 8 (CK8) | 114145561 | 2.40 | 0.0093 | C, N | CO, CA, AP |
| Cytokeratin, type I cytoskeletal 18 (CK18) | 254540068 | 2.33 | 0.0019 | C | CO, CA, AP |
| Prohibitin 1 (PHB1, PHB) | 6679299 | 2.25 | <0.001 | Mi, C, N | TR, MiF |
| Prohibitin 2 (PHB2) | 126723336 | 2.76 | <0.001 | Mi, C, N | TR, MiF |
| Glutathione S-transferase Mu 1 (GSTM1) | 6754084 | 3.52 | <0.001 | C | GM |
| Peroxisomal bifunctional enzyme (EHHADH) | 31541815 | 2.09 | <0.001 | P | LM, FABO, PPARS |
| Thioredoxin-dependent peroxide reductase, mit. (PRDX3) | 6680690 | 2.36 | 0.013 | Mi | ORP, CP, NRA |
| Ornithine aminotransferase, mitochondrial (OAT) | 8393866 | 2.47 | <0.001 | Mi | AAM, AC |
| Group/ Duration | No. Mice | CACHD1+ F/BF | CACHD1+ F/EF | CACHD1+F/MF | Non-BF, EF, MF CACHD1+ F | Total CACHD1+F/Total AF | Total CACHD1−F/Total AF | CACHD1+ HCA/ Total HCA | CACHD1+ HCC/ Total HCC |
|---|---|---|---|---|---|---|---|---|---|
| STZ/18 w | 4 | 2/3 | 2/3 | 7/7 | 0 | 12/13 | 1/13 | 0/0 | 0/0 |
| Incidence (%) | 66.7 | 66.7 | 100 | 0 | 92.3 | 7.7 | 0 | 0 | |
| STAM/18 w | 7 | 30/33 | 60/62 | 107/114 | 25 | 222/234 | 12/234 | 19/19 | 7/7 |
| Incidence (%) | 90.1 | 96.8 | 93.9 | - | 94.9 | 5.1 | 100 | 100 |
| Name | ID | CACHD1kn Huh7/HepG2 | Location | Type | Function |
|---|---|---|---|---|---|
| Protein folding and unfolded protein response | |||||
| Cache domain-containing 1 (CACHD1) | 14285643 | ↓/↓ | U | O | CH |
| Calreticulin (CALR) | 117501 | ↓/↓ | C, EPR | TR | PF, UPR, TR |
| Calumenin (CALU) | 5921197 | ↓/↓ | C, EPR | O | PF, UPR,CPMP |
| Heat shock 70kDa protein 2 (HSPA2) | 1708307 | ↓/↓ | C, EPR | O | UPR |
| Heat shock 70kDa protein 9 (mortalin) (HSPA9) | 21264428 | ↓/↓ | C, EPR | O | UPR |
| Heat shock 70kDa protein 5 (Glu-regul. protein 78kDa) (HSPA5) | 14916999 | ↓/↓ | C, EPR | E | UPR |
| Cytoskeleton organization | |||||
| Cytokeratin 8 (CK8) | 90110027 | ↓/↓ | C | O | CO |
| Cytokeratin 18 (CK18) | 125083 | ↓/↓ | C | O | CO |
| Cytokeratin 19 (CK19) | 311033484 | ↓/↓ | C | O | CO |
| Actin beta like 2 (ACTBL2) | 172046825 | −2.13/−2.05 | N | O | ACO |
| ENAH actin regulator (ENAH) | 48428086 | −2.38/−2.13 | PM | O | ACO |
| Myristoylated alanine-rich protein kinase C substrate (MARCKS) | 76803798 | ↓/↓ | PM | O | ACO |
| Cofilin 1 (non-muscle) (CFL1) | 116848 | ↓/↓ | N | O | ACO |
| MARCKS-like 1 (MARCKSL1) | 1346576 | ↓/↓ | C | O | ACO, Ca T, CP |
| Profilin 1 (PFN1) | 130979 | ↓/↓ | C | O | ACO |
| Tubulin, alpha 1c (TUBA1C) | 20455322 | ↓/↓ | C | O | CO, CD, CDIT |
| Stress response, apoptosis, autophagy | |||||
| Superoxide dismutase 2, mit. (SOD) | 134665 | ↓/↓ | C | E | NRAP, MiF |
| Y box binding protein 1 (YBX1) | 54040030 | ↓/↓ | N | TR | NRAP; NRCS |
| Nucleophosmin (numatrin) (NPM1) | 114762 | −2.2/−2.0 | N | TR | NRAP, CP; CCP |
| Alpha-fetoprotein (AFP) | 120042 | −3.02/−2.75 | ES | T | CPM |
| Nucleolin (NCL) | 90110781 | ↑/↑ | N | O | A |
| Lemur tyrosine kinase 3 (LMTK3) | 117949603 | ↑/↑ | O | K | A, AP |
| Prolyl endopeptidase (PREP) | 215273868 | ↓/↓ | C | P | A |
| Epoxide hydrolase 1, microsomal (xenobiotic) (EPHX1) | 123926 | −2.30/−2.12 | C | P | XM |
| Aspartate aminotransferase 2 (GOT2) | 308153643 | ↓/↓ | C | E | AAM, FAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakehashi, A.; Chariyakornkul, A.; Suzuki, S.; Khuanphram, N.; Tatsumi, K.; Yamano, S.; Fujioka, M.; Gi, M.; Wongpoomchai, R.; Wanibuchi, H. Cache Domain Containing 1 Is a Novel Marker of Non-Alcoholic Steatohepatitis-Associated Hepatocarcinogenesis. Cancers 2021, 13, 1216. https://doi.org/10.3390/cancers13061216
Kakehashi A, Chariyakornkul A, Suzuki S, Khuanphram N, Tatsumi K, Yamano S, Fujioka M, Gi M, Wongpoomchai R, Wanibuchi H. Cache Domain Containing 1 Is a Novel Marker of Non-Alcoholic Steatohepatitis-Associated Hepatocarcinogenesis. Cancers. 2021; 13(6):1216. https://doi.org/10.3390/cancers13061216
Chicago/Turabian StyleKakehashi, Anna, Arpamas Chariyakornkul, Shugo Suzuki, Napaporn Khuanphram, Kumiko Tatsumi, Shotaro Yamano, Masaki Fujioka, Min Gi, Rawiwan Wongpoomchai, and Hideki Wanibuchi. 2021. "Cache Domain Containing 1 Is a Novel Marker of Non-Alcoholic Steatohepatitis-Associated Hepatocarcinogenesis" Cancers 13, no. 6: 1216. https://doi.org/10.3390/cancers13061216
APA StyleKakehashi, A., Chariyakornkul, A., Suzuki, S., Khuanphram, N., Tatsumi, K., Yamano, S., Fujioka, M., Gi, M., Wongpoomchai, R., & Wanibuchi, H. (2021). Cache Domain Containing 1 Is a Novel Marker of Non-Alcoholic Steatohepatitis-Associated Hepatocarcinogenesis. Cancers, 13(6), 1216. https://doi.org/10.3390/cancers13061216