
A Novel Role of Bergamottin in Attenuating Cancer Associated Cachexia by Diverse Molecular Mechanisms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
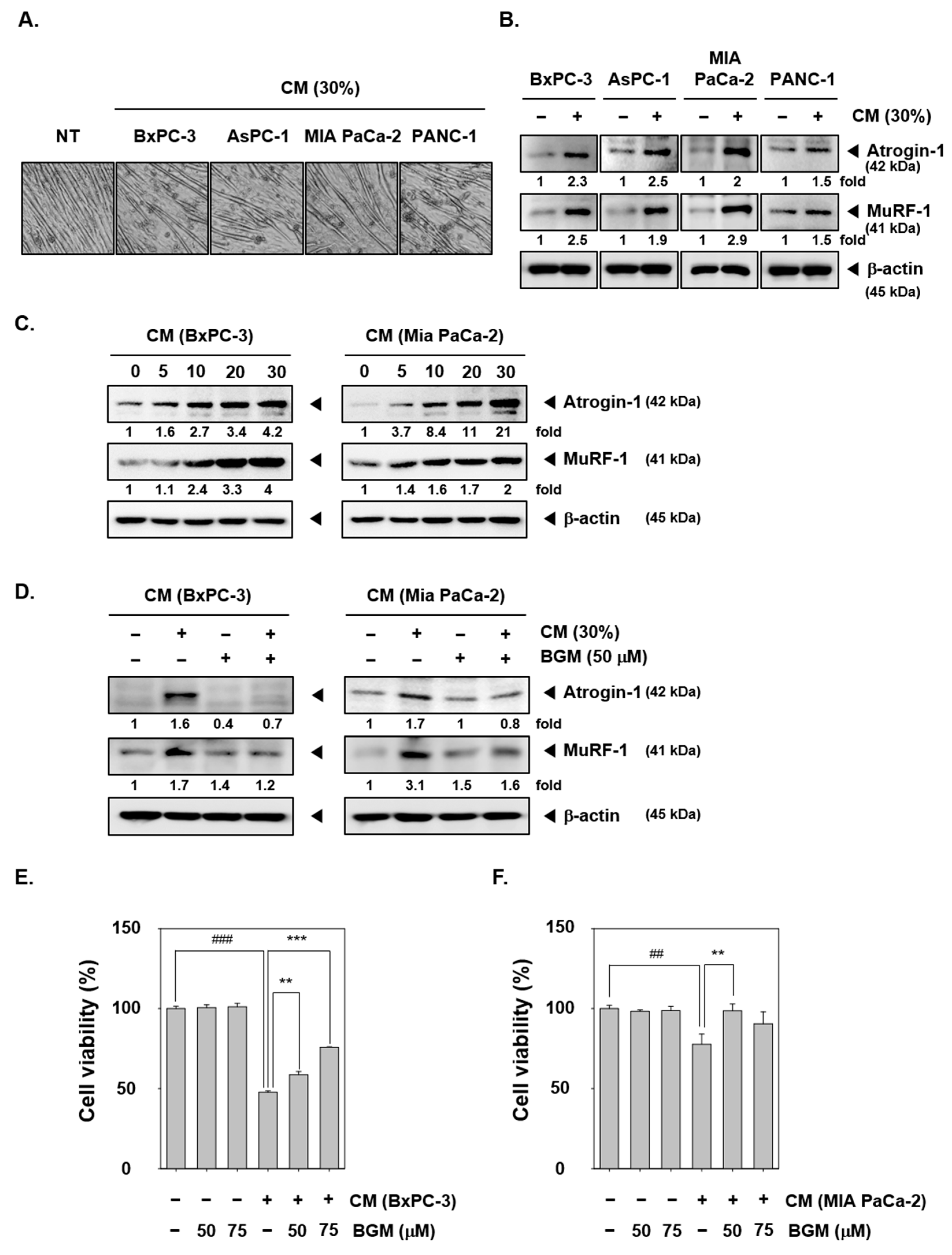
2.1. BGM Suppresses the Pancreatic Cancer Conditioned Media (CM)-Induced Cancer Cachexia in C2C12 Mouse Myoblast Cells In Vitro
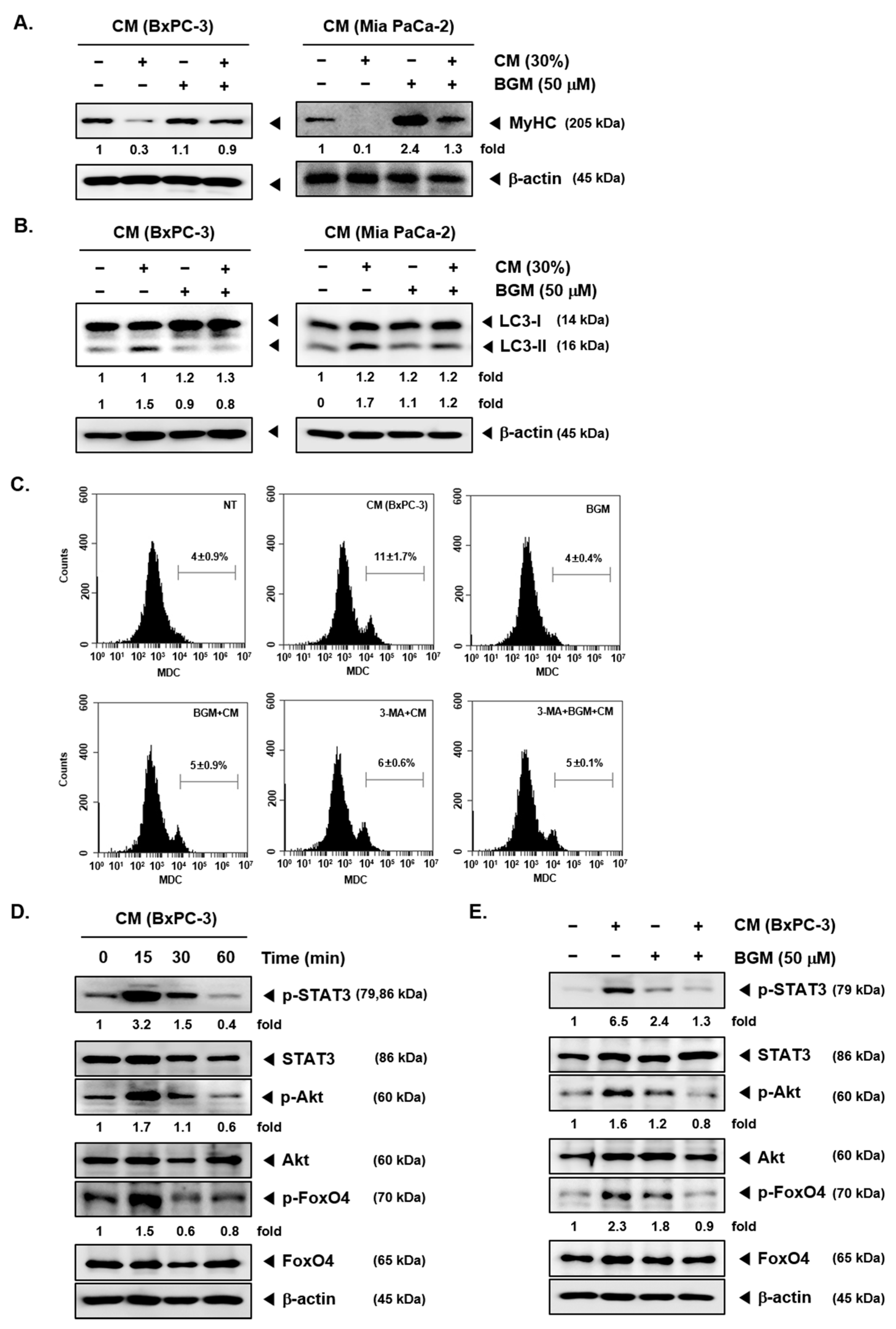
2.2. BGM Can Induce the Myosin Heavy Chain (MyHC) Expression Cancer Cachexia-Induced C2C12 Myotubes In Vitro
2.3. Cancer Cachexia-Induced Autophagy Can Be Suppressed by BGM in C2C12 Myotubes In Vitro
2.4. BGM Inhibits Cachexia-Induced Inflammatory Signaling Factors in C2C12 Myotubes In Vitro
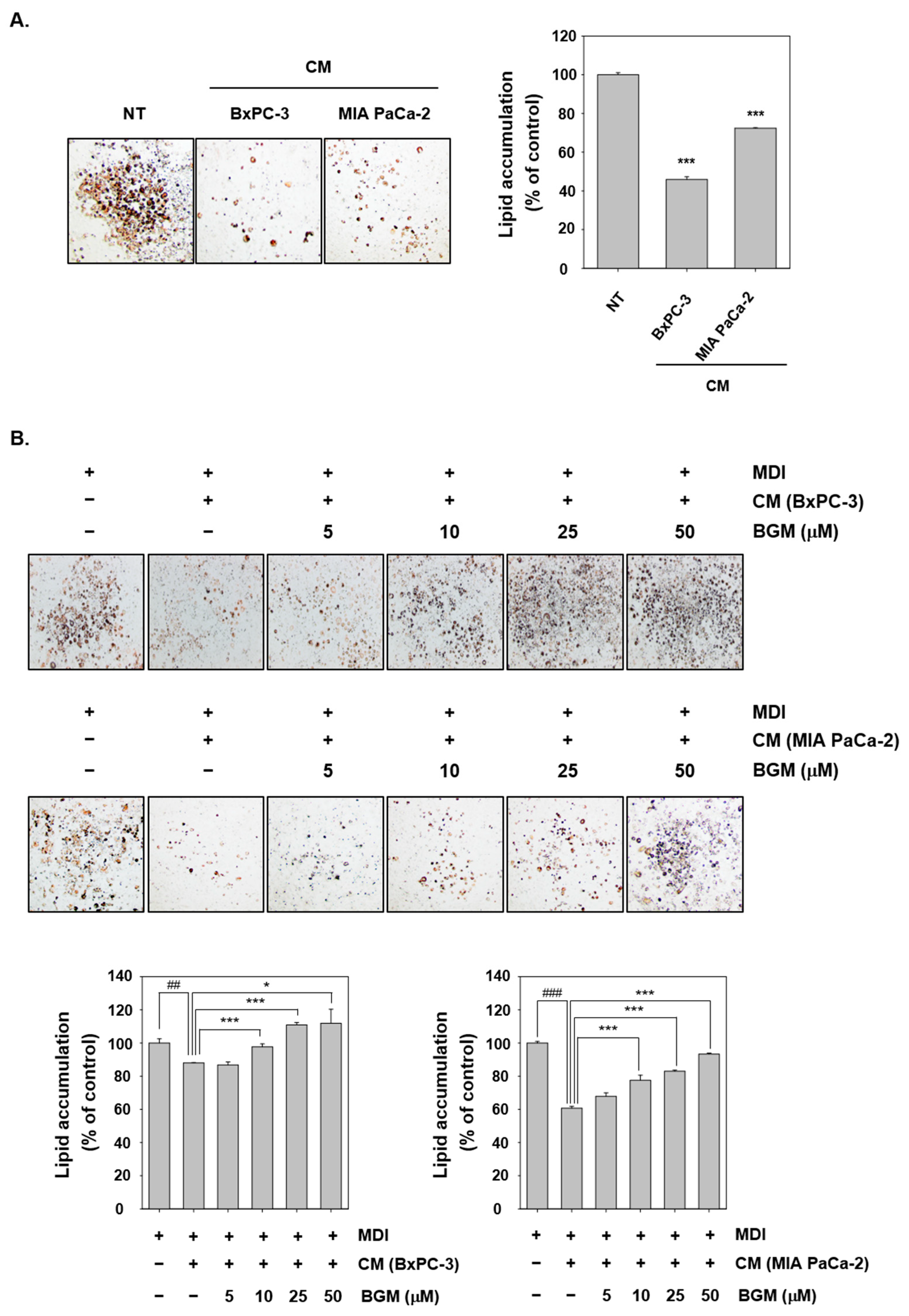
2.5. Cancer Cachexia Is Induced by CM in 3T3L1 Mouse Fibroblast Cells In Vitro
2.6. BGM Can Induce Adipogenesis- and Differentiation-Related Factors in 3T3L1 Cells In Vitro
2.7. BGM Inhibits Cachexia-Induced Inflammatory Signaling Factors in 3T3L1 Adipocytes In Vitro
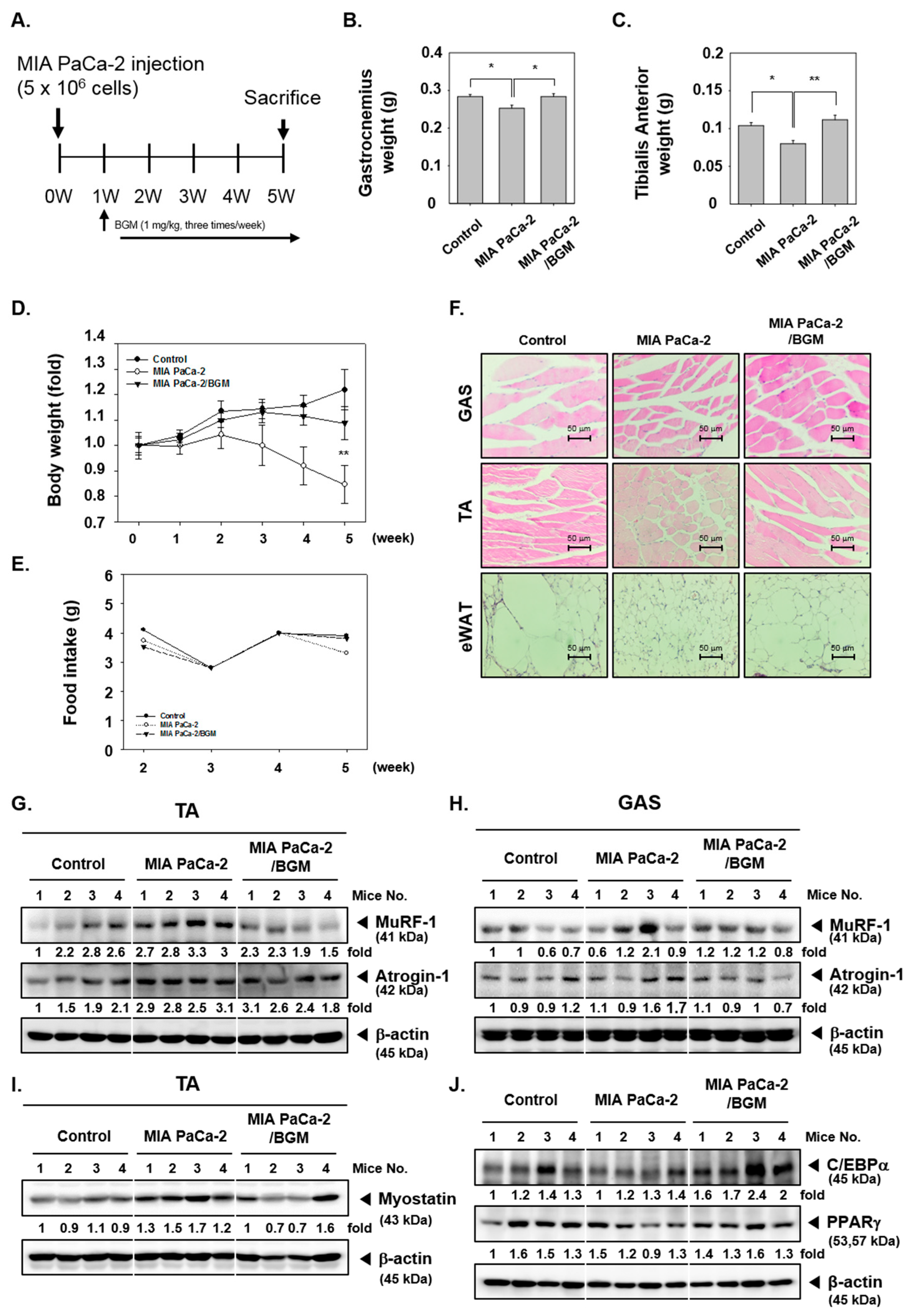
2.8. BGM Suppresses Cancer Cachexia and Modulates the Expression of Skeletal Muscle- and Adipogenesis-Related Factors In Vivo in a Xenograft Mouse Model
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Culture Conditions
4.3. MTT Assay
4.4. Western Blot Analysis
4.5. MDC Staining
4.6. Oil Red O Staining
4.7. Animals
4.8. Experimental Protocol
4.9. Western Blot Analysis of Tumor Tissues
4.10. H&E Staining
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BGM | Bergamottin |
| STAT3 | signal transducer and activator of transcription 3 |
| c/w | cell per well |
| FBS | fetal bovine serum |
| HRP | horseradish peroxidase |
| H&E | Hematoxylin and Eosin |
| i.p. | intraperitoneal |
| CM | Conditioned media |
| MyHC | myosin heavy chain |
| NT | nontreated |
| P/S | penicillin–streptomycin |
| ZAG | Zinc-α2 glycoprotein |
| HSL | hormone-sensitive lipase |
| SFM | serum-free media |
| TA | Tibialis anterior |
| GAS | gastrocnemius |
| eWAT | epididymal white adipose tissue |
References
- Dhanapal, R.; Saraswathi, T.; Govind, R.N. Cancer cachexia. J. Oral Maxillofac. Pathol. 2011, 15, 257–260. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, K.C.; Voss, A.C.; Hustead, D.S.; Cancer Cachexia Study, G. Definition of cancer cachexia: Effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am. J. Clin. Nutr. 2006, 83, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Kiss, N.; Hodgson, B.; Crowe, T.C.; Walsh, A.D. Associations between nutritional status, weight loss, radiotherapy treatment toxicity and treatment outcomes in gastrointestinal cancer patients. Clin. Nutr. 2011, 30, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Theologides, A. Cancer cachexia. Cancer 1979, 43, 2004–2012. [Google Scholar] [CrossRef]
- Mitch, W.E.; Goldberg, A.L. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N. Engl. J. Med. 1996, 335, 1897–1905. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein turnover in skeletal muscle. II. Effects of denervation and cortisone on protein catabolism in skeletal muscle. J. Biol. Chem. 1969, 244, 3223–3229. [Google Scholar] [CrossRef]
- Mitch, W.E.; Medina, R.; Grieber, S.; May, R.C.; England, B.K.; Price, S.R.; Bailey, J.L.; Goldberg, A.L. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J. Clin. Investig. 1994, 93, 2127–2133. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [Green Version]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Labeit, S.; Kohl, C.H.; Witt, C.C.; Labeit, D.; Jung, J.; Granzier, H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J. Biomed. Biotechnol. 2010, 2010, 693741. [Google Scholar] [CrossRef] [Green Version]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Tsoli, M.; Robertson, G. Cancer cachexia: Malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 2013, 24, 174–183. [Google Scholar] [CrossRef]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Inacio Pinto, N.; Carnier, J.; Oyama, L.M.; Otoch, J.P.; Alcantara, P.S.; Tokeshi, F.; Nascimento, C.M. Cancer as a Proinflammatory Environment: Metastasis and Cachexia. Mediat. Inflamm. 2015, 2015, 791060. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Shao, J. Adiponectin and energy homeostasis. Rev. Endocr. Metab. Disord. 2014, 15, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Tsuchiya, H.; Hama, S.; Kajimoto, K.; Kogure, K. Resistin affects lipid metabolism during adipocyte maturation of 3T3-L1 cells. FEBS J. 2013, 280, 5884–5895. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Hasanpourghadi, M.; Looi, C.Y.; Pandurangan, A.K.; Sethi, G.; Wong, W.F.; Mustafa, M.R. Phytometabolites Targeting the Warburg Effect in Cancer Cells: A Mechanistic Review. Curr. Drug Targets 2017, 18, 1086–1094. [Google Scholar] [CrossRef]
- Deorukhkar, A.; Krishnan, S.; Sethi, G.; Aggarwal, B.B. Back to basics: How natural products can provide the basis for new therapeutics. Expert Opin. Investig. Drugs 2007, 16, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Warrier, S.; Merarchi, M.; Arfuso, F.; Kumar, A.P.; Bishayee, A. Pro-Apoptotic and Anti-Cancer Properties of Diosgenin: A Comprehensive and Critical Review. Nutrients 2018, 10, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. 2015, 240, 760–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef]
- Dolan, L.C.; Matulka, R.A.; Burdock, G.A. Naturally occurring food toxins. Toxins 2010, 2, 2289–2332. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, D.J. Dietary exposure to furocoumarins. Regul. Toxicol. Pharmacol. 1991, 14, 261–272. [Google Scholar] [CrossRef]
- Hung, W.L.; Suh, J.H.; Wang, Y. Chemistry and health effects of furanocoumarins in grapefruit. J. Food Drug Anal. 2017, 25, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.H.; Arfuso, F.; Sethi, G.; Ahn, K.S. Pharmacological Utilization of Bergamottin, Derived from Grapefruits, in Cancer Prevention and Therapy. Int. J. Mol. Sci. 2018, 19, 4048. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lee, J.H.; Sethi, G.; Kim, C.; Baek, S.H.; Nam, D.; Chung, W.S.; Kim, S.H.; Shim, B.S.; Ahn, K.S. Bergamottin, a natural furanocoumarin obtained from grapefruit juice induces chemosensitization and apoptosis through the inhibition of STAT3 signaling pathway in tumor cells. Cancer Lett. 2014, 354, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Nam, D.; Um, J.Y.; Jung, S.H.; Ahn, K.S. Bergamottin Inhibits Adipogenesis in 3T3-L1 Cells and Weight Regulation in Diet-Induced Obese Mice. Am. J. Chin. Med. 2018, 46, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Hatano, T.; Taketomi, A.; Kawabata, M.; Nakabayashi, T. Bergamottin Promotes Adipocyte Differentiation and Inhibits Tumor Necrosis Factor-alpha-induced Inflammatory Cytokines Induction in 3T3-L1 Cells. Yakugaku Zasshi 2017, 137, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Solheim, T.S.; Laird, B.J.A.; Balstad, T.R.; Stene, G.B.; Bye, A.; Johns, N.; Pettersen, C.H.; Fallon, M.; Fayers, P.; Fearon, K.; et al. A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Solheim, T.S.; Laird, B.J.A.; Balstad, T.R.; Bye, A.; Stene, G.; Baracos, V.; Strasser, F.; Griffiths, G.; Maddocks, M.; Fallon, M.; et al. Cancer cachexia: Rationale for the MENAC (Multimodal-Exercise, Nutrition and Anti-inflammatory medication for Cachexia) trial. BMJ Support. Palliat. Care 2018, 8, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, Y.L.; Fu, C.Y.; Wang, J.; Chen, S.Y.; Yao, J.; Lai, S.J. Expression of MyHC genes, composition of muscle fiber type and their association with intramuscular fat, tenderness in skeletal muscle of Simmental hybrids. Mol. Biol. Rep. 2014, 41, 833–840. [Google Scholar] [CrossRef]
- Bing, C.; Trayhurn, P. New insights into adipose tissue atrophy in cancer cachexia. Proc. Nutr. Soc. 2009, 68, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Stubbins, R.; Bernicker, E.H.; Quigley, E.M.M. Cancer cachexia: A multifactoral disease that needs a multimodal approach. Curr. Opin. Gastroenterol. 2020, 36, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.F.; Rohm, M.; Herzig, S.; Diaz, M.B. Cancer Cachexia: More than Skeletal Muscle Wasting. Trends Cancer 2018, 4, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Investig. 2004, 114, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Ballaro, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argiles, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 54, pp. 28–41. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-J.; Yan, H.; Han, Y.-L.; Wan, L.-L.; Li, J.; Huang, J.-L.; Lu, J.; Chen, P.-G.; Gan, R.; Guo, C. Selumetinib Attenuates Skeletal Muscle Wasting in Murine Cachexia Model through ERK Inhibition and AKT Activation. Mol. Cancer Ther. 2017, 16, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.A.; Sandesara, P.B.; Senf, S.M.; Judge, A.R. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 2012, 26, 987–1000. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Lee, J.H.; Nam, D.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Um, J.Y.; Sethi, G.; Ahn, K.S. Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade. Front. Pharmacol. 2018, 9, 531. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386–6405. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Shanmugam, M.K.; Narula, A.S.; Kim, C.; Lee, J.H.; Namjoshi, O.A.; Blough, B.E.; Sethi, G.; Ahn, K.S. Oxymatrine Attenuates Tumor Growth and Deactivates STAT5 Signaling in a Lung Cancer Xenograft Model. Cancers 2019, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Cho, S.K.; Kapoor, S.; Kumar, A.; Vali, S.; Abbasi, T.; Kim, S.H.; Sethi, G.; Ahn, K.S. β-Caryophyllene oxide inhibits constitutive and inducible STAT3 signaling pathway through induction of the SHP-1 protein tyrosine phosphatase. Mol. Carcinog. 2014, 53, 793–806. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.Y.; Ko, J.-H.; Um, J.-Y.; Sethi, G.; Ahn, K.S. A Novel Role of Bergamottin in Attenuating Cancer Associated Cachexia by Diverse Molecular Mechanisms. Cancers 2021, 13, 1347. https://doi.org/10.3390/cancers13061347
Jung YY, Ko J-H, Um J-Y, Sethi G, Ahn KS. A Novel Role of Bergamottin in Attenuating Cancer Associated Cachexia by Diverse Molecular Mechanisms. Cancers. 2021; 13(6):1347. https://doi.org/10.3390/cancers13061347
Chicago/Turabian StyleJung, Young Yun, Jeong-Hyeon Ko, Jae-Young Um, Gautam Sethi, and Kwang Seok Ahn. 2021. "A Novel Role of Bergamottin in Attenuating Cancer Associated Cachexia by Diverse Molecular Mechanisms" Cancers 13, no. 6: 1347. https://doi.org/10.3390/cancers13061347