
Large Scale Molecular Studies of Pituitary Neuroendocrine Tumors: Novel Markers, Mechanisms and Translational Perspectives

Abstract
:Simple Summary
Abstract
1. Introduction
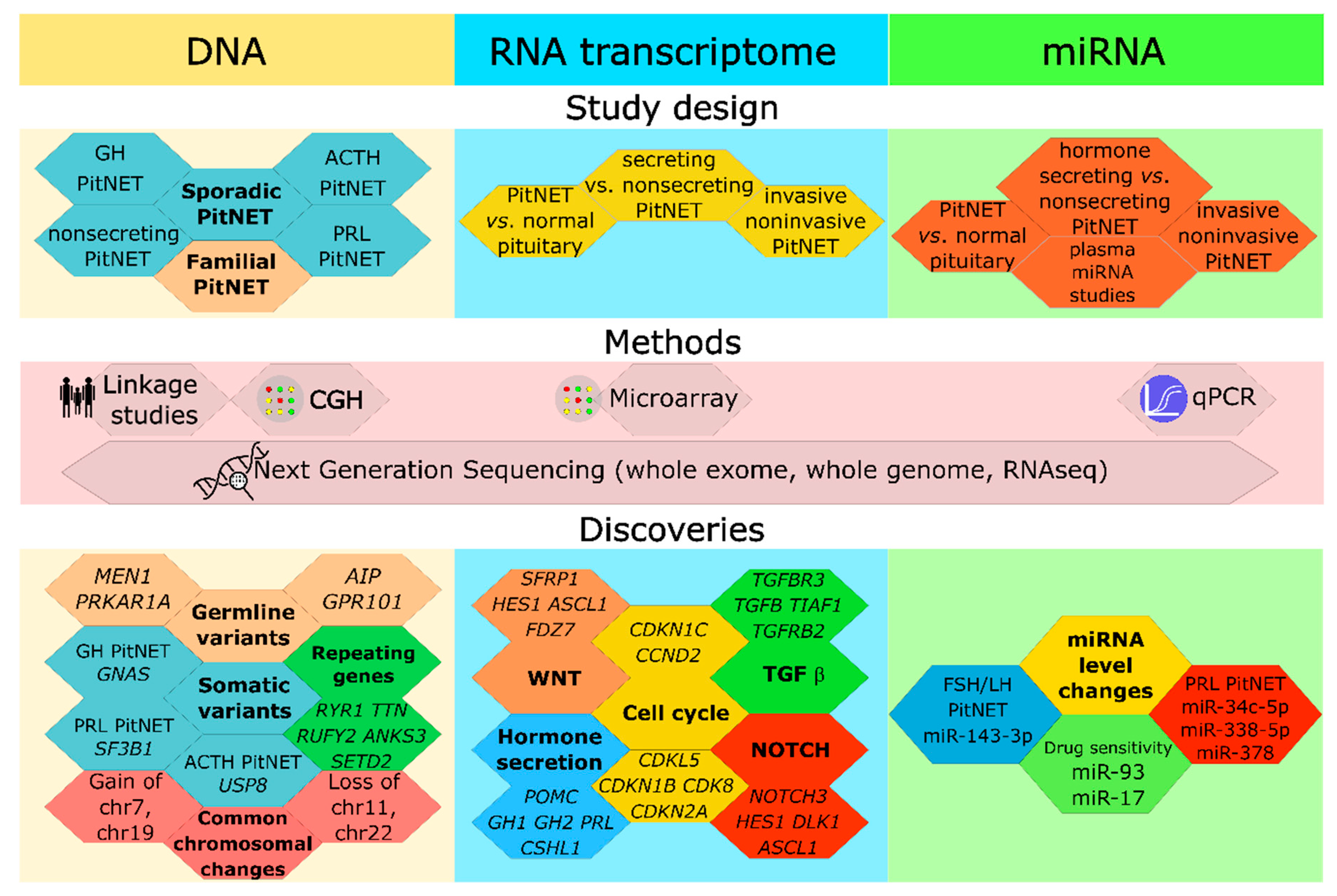
Review Terminology and Methodology
2. The Landscape of Genomic Alterations in PitNETs
2.1. Familial PitNETs
2.2. Sporadic PitNET
2.2.1. GNAS
2.2.2. USP8
2.2.3. SF3B1
2.2.4. MEN1
2.3. Genome Changes in PitNETs
2.4. WES, WGS Somatic Mutation Studies
2.5. Recurrent Genes with Somatic Variants
3. Large-Scale Transcriptomics of PitNETs
3.1. Microarray-Based Approach
3.2. Whole Transcriptome Sequencing and Pangenomic Classification of PitNETs
3.3. Overlapping Transcriptome Markers
4. Micro RNAs in PitNET Pathogenesis
Circulating miRNAs
5. Regulatory Effects of Long Non-Coding RNA in PitNETs
6. Translational Perspective
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Daly, A.F.; Rixhon, M.; Adam, C.; Dempegioti, A.; Tichomirowa, M.A.; Beckers, A. High prevalence of pituitary adenomas: A cross-sectional study in the province of Liège, Belgium. J. Clin. Endocrinol. Metab. 2006, 91, 4769–4775. [Google Scholar] [CrossRef]
- Schneider, H.J.; Sievers, C.; Saller, B.; Wittchen, H.U.; Stalla, G.K. High prevalence of biochemical acromegaly in primary care patients with elevated IGF-1 levels. Clin. Endocrinol. (Oxf.) 2008, 69, 432–435. [Google Scholar] [CrossRef]
- Rosario, P.W.; Calsolari, M.R. Screening for acromegaly by application of a simple questionnaire evaluating the enlargement of extremities in adult patients seen at primary health care units. Pituitary 2012, 15, 179–183. [Google Scholar] [CrossRef]
- Ntali, G.; Wass, J.A. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary 2018, 21, 111–118. [Google Scholar] [CrossRef]
- Aflorei, E.D.; Korbonits, M. Epidemiology and etiopathogenesis of pituitary adenomas. J. Neurooncol. 2014, 117, 379–394. [Google Scholar] [CrossRef]
- Daly, A.F.; Tichomirowa, M.A.; Beckers, A. The epidemiology and genetics of pituitary adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 543–554. [Google Scholar] [CrossRef]
- Asa, S.L.; Ezzat, S. The pathogenesis of pituitary tumours. Nat. Rev. Cancer 2002, 2, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S.; Bronstein, M.D.; Chanson, P.; Klibanski, A.; Casanueva, F.F.; Wass, J.A.H.; Strasburger, C.J.; Luger, A.; Clemmons, D.R.; Giustina, A. A Consensus Statement on acromegaly therapeutic outcomes. Nat. Rev. Endocrinol. 2018, 14, 552–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, J.B.; Ribeiro-Oliveira, A.J.; Soares, B.S. Non-Functioning Pituitary Adenomas; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; Endotext [Internet]: South Dartmouth, MA, USA; MDText.com, Inc., 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK534880/ (accessed on 18 March 2021).
- Öberg, K.; Lamberts, S.W.J. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumours: Past, present and future. Endocr. Relat. Cancer 2016, 23, R551–R566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque-Ramírez, M.; Paramo, C.; Varela da Costa, C.; García-Mayor, R.V. Cost of management of invasive growth hormone-secreting macroadenoma. J. Endocrinol. Investig. 2007, 30, 541–545. [Google Scholar] [CrossRef]
- Didoni, G.; Grottol, S.; Gasco, V.; Battistini, M.; Ferone, D.; Giusti, M.; Ragazzoni, F.; Ruffo, P.; Ghigo, E.; Minuto, F. Cost-of-illness study in acromegalic patients in Italy. J. Endocrinol. Investig. 2004, 27, 1034–1039. [Google Scholar] [CrossRef]
- Caimari, F.; Korbonits, M. Novel Genetic Causes of Pituitary Adenomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5030–5042. [Google Scholar] [CrossRef] [Green Version]
- Vandeva, S.; Daly, A.F.; Petrossians, P.; Zacharieva, S.; Beckers, A. Somatic and germline mutations in the pathogenesis of pituitary adenomas. Eur. J. Endocrinol. 2019, 181, R235–R254. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-W.; Wang, Y.-Q.; Shu, H.-S. MiR-16 inhibits pituitary adenoma cell proliferation via the suppression of ERK/MAPK signal pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Gruppetta, M.; Formosa, R.; Falzon, S.; Ariff Scicluna, S.; Falzon, E.; Degeatano, J.; Vassallo, J. Expression of cell cycle regulators and biomarkers of proliferation and regrowth in human pituitary adenomas. Pituitary 2017, 20, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Paragliola, R.M.; Corsello, S.M.; Salvatori, R. Somatostatin receptor ligands in acromegaly: Clinical response and factors predicting resistance. Pituitary 2017, 20, 109–115. [Google Scholar] [CrossRef]
- Colao, A.; Auriemma, R.S.; Pivonello, R. The effects of somatostatin analogue therapy on pituitary tumor volume in patients with acromegaly. Pituitary 2016, 19, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadelha, M.R.; Wildemberg, L.E.; Bronstein, M.D.; Gatto, F.; Ferone, D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary 2017, 20, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Newey, P.J.; Nesbit, M.A.; Rimmer, A.J.; Head, R.A.; Gorvin, C.M.; Attar, M.; Gregory, L.; Wass, J.A.H.; Buck, D.; Karavitaki, N.; et al. Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab. 2013, 98, E796–E800. [Google Scholar] [CrossRef] [Green Version]
- Välimäki, N.; Demir, H.; Pitkänen, E.; Kaasinen, E.; Karppinen, A.; Kivipelto, L.; Schalin-Jäntti, C.; Aaltonen, L.A.; Karhu, A. Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2015, 100, 3918–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.-J.; Reitman, Z.J.; Ma, Z.-Y.; Chen, J.-H.; Zhang, Q.-L.; Shou, X.-F.; Huang, C.-X.; Wang, Y.-F.; Li, S.-Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Bi, W.L.; Horowitz, P.; Greenwald, N.F.; Abedalthagafi, M.; Agarwalla, P.K.; Gibson, W.J.; Mei, Y.; Schumacher, S.E.; Ben-David, U.; Chevalier, A.; et al. Landscape of Genomic Alterations in Pituitary Adenomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1841–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Kim, J.H. Transcriptome Analysis Identifies an Attenuated Local Immune Response in Invasive Nonfunctioning Pituitary Adenomas. Endocrinol. Metab. (Seoul, Korea) 2019, 34, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.-L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134.e5. [Google Scholar] [CrossRef] [PubMed]
- Salomon, M.P.; Wang, X.; Marzese, D.M.; Hsu, S.C.; Nelson, N.; Zhang, X.; Matsuba, C.; Takasumi, Y.; Ballesteros-Merino, C.; Fox, B.A.; et al. The Epigenomic Landscape of Pituitary Adenomas Reveals Specific Alterations and Differentiates Among Acromegaly, Cushing’s Disease and Endocrine-Inactive Subtypes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4126–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falch, C.M.; Sundaram, A.Y.M.; Øystese, K.A.; Normann, K.R.; Lekva, T.; Silamikelis, I.; Eieland, A.K.; Andersen, M.; Bollerslev, J.; Olarescu, N.C. Gene expression profiling of fast- and slow-growing non-functioning gonadotroph pituitary adenomas. Eur. J. Endocrinol. 2018, 178, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.R.; Yan, L.; Liu, Y.T.; Cao, L.; Guo, Y.H.; Zhang, Y.; Yao, H.; Cai, L.; Shang, H.B.; Rui, W.W.; et al. Inhibition of mTORC1 by lncRNA H19 via disrupting 4E-BP1/Raptor interaction in pituitary tumours. Nat. Commun. 2018, 9, 4624. [Google Scholar] [CrossRef] [Green Version]
- Xing, W.; Qi, Z.; Huang, C.; Zhang, N.; Zhang, W.; Li, Y.; Qiu, M.; Fang, Q.; Hui, G. Genome-wide identification of lncRNAs and mRNAs differentially expressed in non-functioning pituitary adenoma and construction of an lncRNA-mRNA co-expression network. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Chen, L.; Hu, X.; Tang, J.; He, L.; Hu, J.; Fei, F.; Wang, Q. Next-generation sequencing of microRNAs reveals a unique expression pattern in different types of pituitary adenomas. Endocr. J. 2019, 66, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, H.; Inoshita, N. New WHO classification of pituitary adenomas (4th edition): Assessment of pituitary transcription factors and the prognostic histological factors. Brain Tumor Pathol. 2018, 35, 57–61. [Google Scholar] [CrossRef]
- Lopes, M.B.S. The 2017 World Health Organization classification of tumors of the pituitary gland: A summary. Acta Neuropathol. 2017, 134, 521–535. [Google Scholar] [CrossRef]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef]
- Daniely, M.; Aviram, A.; Adams, E.F.; Buchfelder, M.; Barkai, G.; Fahlbusch, R.; Goldman, B.; Friedman, E. Comparative genomic hybridization analysis of nonfunctioning pituitary tumors. J. Clin. Endocrinol. Metab. 1998, 83, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Peculis, R.; Mandrika, I.; Petrovska, R.; Dortane, R.; Megnis, K.; Nazarovs, J.; Balcere, I.; Stukens, J.; Konrade, I.; Pirags, V.; et al. Pituispheres Contain Genetic Variants Characteristic to Pituitary Adenoma Tumor Tissue. Front. Endocrinol. (Lausanne) 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, L.B.; Hedley-Whyte, E.T.; Pulaski, K.; Seizinger, B.R.; Martuza, R.L. Clonal origin of pituitary adenomas. J. Neurosurg. 1990, 73, 731–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaid, M.; Korbonits, M. Genetics of pituitary adenomas. Neurol. India 2017, 65, 577–587. [Google Scholar] [CrossRef]
- Couldwell, W.T.; Cannon-Albright, L. A heritable predisposition to pituitary tumors. Pituitary 2010, 13, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Larsson, C.; Julier, C.; Byström, C.; Skogseid, B.; Wells, S.; Oberg, K.; Carlson, M.; Taggart, T.; O’Connell, P. Localization of the genetic defect in multiple endocrine neoplasia type 1 within a small region of chromosome 11. Am. J. Hum. Genet. 1989, 44, 751–755. [Google Scholar]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Forlino, A.; Vetro, A.; Garavelli, L.; Ciccone, R.; London, E.; Stratakis, C.A.; Zuffardi, O. PRKACB and Carney complex. N. Engl. J. Med. 2014, 370, 1065–1067. [Google Scholar] [CrossRef]
- Daly, A.F.; Jaffrain-Rea, M.-L.; Ciccarelli, A.; Valdes-Socin, H.; Rohmer, V.; Tamburrano, G.; Borson-Chazot, C.; Estour, B.; Ciccarelli, E.; Brue, T.; et al. Clinical characterization of familial isolated pituitary adenomas. J. Clin. Endocrinol. Metab. 2006, 91, 3316–3323. [Google Scholar] [CrossRef] [Green Version]
- Xekouki, P.; Szarek, E.; Bullova, P.; Giubellino, A.; Quezado, M.; Mastroyannis, S.A.; Mastorakos, P.; Wassif, C.A.; Raygada, M.; Rentia, N.; et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J. Clin. Endocrinol. Metab. 2015, 100, E710–E719. [Google Scholar] [CrossRef] [PubMed]
- De Kock, L.; Sabbaghian, N.; Plourde, F.; Srivastava, A.; Weber, E.; Bouron-Dal Soglio, D.; Hamel, N.; Choi, J.H.; Park, S.-H.; Deal, C.L.; et al. Pituitary blastoma: A pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014, 128, 111–122. [Google Scholar] [CrossRef]
- Bengtsson, D.; Joost, P.; Aravidis, C.; Askmalm Stenmark, M.; Backman, A.-S.; Melin, B.; von Salomé, J.; Zagoras, T.; Gebre-Medhin, S.; Burman, P. Corticotroph Pituitary Carcinoma in a Patient With Lynch Syndrome (LS) and Pituitary Tumors in a Nationwide LS Cohort. J. Clin. Endocrinol. Metab. 2017, 102, 3928–3932. [Google Scholar] [CrossRef]
- Uraki, S.; Ariyasu, H.; Doi, A.; Furuta, H.; Nishi, M.; Sugano, K.; Inoshita, N.; Nakao, N.; Yamada, S.; Akamizu, T. Atypical pituitary adenoma with MEN1 somatic mutation associated with abnormalities of DNA mismatch repair genes; MLH1 germline mutation and MSH6 somatic mutation. Endocr. J. 2017, 64, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, R.A.; Mendonca, B.B.; Fragoso, M.C.B.V.; Soares, I.C.; Almeida, M.Q.; Moraes, M.B.; Lourenço, D.M.J.; Alves, V.A.F.; Bronstein, M.D.; Toledo, S.P.A. Isolated familial somatotropinoma: 11q13-loh and gene/protein expression analysis suggests a possible involvement of aip also in non-pituitary tumorigenesis. Clinics (Sao Paulo) 2010, 65, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Höfler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef] [Green Version]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.-H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Klibanski, A. Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol. Cell. Endocrinol. 2014, 386, 16–33. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Li, Z.; Wang, Y.; Mao, Y.; Shen, M.; Zhang, Q.; Li, S.; Zhou, L.; Shou, X.; Chen, J.; et al. Common variants at 10p12.31, 10q21.1 and 13q12.13 are associated with sporadic pituitary adenoma. Nat. Genet. 2015, 47, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taboada, G.F.; Tabet, A.L.O.; Naves, L.A.; de Carvalho, D.P.; Gadelha, M.R. Prevalence of gsp oncogene in somatotropinomas and clinically non-functioning pituitary adenomas: Our experience. Pituitary 2009, 12, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45, D635–D642. [Google Scholar] [CrossRef]
- Freda, P.U.; Chung, W.K.; Matsuoka, N.; Walsh, J.E.; Kanibir, M.N.; Kleinman, G.; Wang, Y.; Bruce, J.N.; Post, K.D. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007, 10, 275–282. [Google Scholar] [CrossRef]
- Patten, J.L.; Johns, D.R.; Valle, D.; Eil, C.; Gruppuso, P.A.; Steele, G.; Smallwood, P.M.; Levine, M.A. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N. Engl. J. Med. 1990, 322, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tang, D.; Deng, J.; Su, C. Detection of gsp oncogene in growth hormone-secreting pituitary adenomas and the study of clinical characteristics of acromegalic patients with gsp-positive pituitary tumors. Chin. Med. J. (Engl.) 1998, 111, 891–894. [Google Scholar]
- Riminucci, M.; Collins, M.T.; Lala, R.; Corsi, A.; Matarazzo, P.; Gehron Robey, P.; Bianco, P. An R201H activating mutation of the GNAS1 (Gsalpha) gene in a corticotroph pituitary adenoma. Mol. Pathol. 2002, 55, 58–60. [Google Scholar] [CrossRef]
- Hage, M.; Viengchareun, S.; Brunet, E.; Villa, C.; Pineau, D.; Bouligand, J.; Teglas, J.-P.; Adam, C.; Parker, F.; Lombès, M.; et al. Genomic Alterations and Complex Subclonal Architecture in Sporadic GH-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2018, 103, 1929–1939. [Google Scholar] [CrossRef]
- Vallar, L.; Spada, A.; Giannattasio, G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 1987, 330, 566–568. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Ramkissoon, S.H.; Abedalthagafi, M.; Coy, S.M.; Ligon, K.L.; Mei, Y.; MacConaill, L.; Ducar, M.; Min, L.; et al. Clinical Identification of Oncogenic Drivers and Copy-Number Alterations in Pituitary Tumors. Endocrinology 2017, 158, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Berlin, I.; Higginbotham, K.M.; Dise, R.S.; Sierra, M.I.; Nash, P.D. The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 2010, 285, 37895–37908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferraù, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Faucz, F.R.; Wildemberg, L.E.; Assié, G.; Beschorner, R.; et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Inoshita, N.; Kawaguchi, K.; Ibrahim Ardisasmita, A.; Suzuki, H.; Fukuhara, N.; Okada, M.; Nishioka, H.; Takeuchi, Y.; Komada, M.; et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur. J. Endocrinol. 2016, 174, 213–226. [Google Scholar] [CrossRef]
- Han, J.; Tian, Y.; Yu, L.; Zhang, Q.; Xu, X.; Zhang, Y.; Wang, J.; Ma, Z.; Bian, J.; Luo, C.; et al. Discovery of novel USP8 inhibitors via Ubiquitin-Rho-110 fluorometric assay based high throughput screening. Bioorg. Chem. 2020, 101, 103962. [Google Scholar] [CrossRef]
- Sun, J.; Shen, D.; Zheng, Y.; Ren, H.; Liu, H.; Chen, X.; Gao, Y. USP8 Inhibitor Suppresses HER-2 Positive Gastric Cancer Cell Proliferation and Metastasis via the PI3K/AKT Signaling Pathway. Onco Targets Ther. 2020, 13, 9941–9952. [Google Scholar] [CrossRef]
- Li, C.; Xie, W.; Rosenblum, J.S.; Zhou, J.; Guo, J.; Miao, Y.; Shen, Y.; Wang, H.; Gong, L.; Li, M.; et al. Somatic SF3B1 hotspot mutation in prolactinomas. Nat. Commun. 2020, 11, 2506. [Google Scholar] [CrossRef]
- Cuny, T.; Pertuit, M.; Sahnoun-Fathallah, M.; Daly, A.; Occhi, G.; Odou, M.F.; Tabarin, A.; Nunes, M.L.; Delemer, B.; Rohmer, V.; et al. Genetic analysis in young patients with sporadic pituitary macroadenomas: Besides AIP don’t forget MEN1 genetic analysis. Eur. J. Endocrinol. 2013, 168, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Peculis, R.; Balcere, I.; Rovite, V.; Megnis, K.; Valtere, A.; Stukens, J.; Arnicane, L.; Nikitina-Zake, L.; Lejnieks, A.; Pirags, V.; et al. Polymorphisms in MEN1 and DRD2 genes are associated with the occurrence and characteristics of pituitary adenomas. Eur. J. Endocrinol. 2016, 175, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Paetau, A.; Aalto, Y.; Välimäki, M.; Sane, T.; Poranen, A.; Castresana, J.S.; Knuutila, S. Gain of chromosome 3 and loss of 13q are frequent alterations in pituitary adenomas. Cancer Genet. Cytogenet. 2001, 128, 97–103. [Google Scholar] [CrossRef]
- Hui, A.B.; Pang, J.C.; Ko, C.W.; Ng, H.K. Detection of chromosomal imbalances in growth hormone-secreting pituitary tumors by comparative genomic hybridization. Hum. Pathol. 1999, 30, 1019–1023. [Google Scholar] [CrossRef]
- Harada, K.; Nishizaki, T.; Ozaki, S.; Kubota, H.; Harada, K.; Okamura, T.; Ito, H.; Sasaki, K. Cytogenetic alterations in pituitary adenomas detected by comparative genomic hybridization. Cancer Genet. Cytogenet. 1999, 112, 38–41. [Google Scholar] [CrossRef]
- Trautmann, K.; Thakker, R.V.; Ellison, D.W.; Ibrahim, A.; Lees, P.D.; Harding, B.; Fischer, C.; Popp, S.; Bartram, C.R.; Jauch, A. Chromosomal aberrations in sporadic pituitary tumors. Int. J. Cancer 2001, 91, 809–814. [Google Scholar] [CrossRef]
- De Sousa, S.M.C.; Wang, P.P.S.; Santoreneos, S.; Shen, A.; Yates, C.J.; Babic, M.; Eshraghi, L.; Feng, J.; Koszyca, B.; Roberts-Thomson, S.; et al. The Genomic Landscape of Sporadic Prolactinomas. Endocr. Pathol. 2019, 30, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, S.; Horiguchi, K.; Tosaka, M.; Yamada, S.; Yamada, M. Whole-Exome Sequencing Study of Thyrotropin-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2017, 102, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Rostomyan, L.; Daly, A.F.; Petrossians, P.; Nachev, E.; Lila, A.R.; Lecoq, A.-L.; Lecumberri, B.; Trivellin, G.; Salvatori, R.; Moraitis, A.G.; et al. Clinical and genetic characterization of pituitary gigantism: An international collaborative study in 208 patients. Endocr. Relat. Cancer 2015, 22, 745–757. [Google Scholar] [CrossRef]
- Lan, X.; Gao, H.; Wang, F.; Feng, J.; Bai, J.; Zhao, P.; Cao, L.; Gui, S.; Gong, L.; Zhang, Y. Whole-exome sequencing identifies variants in invasive pituitary adenomas. Oncol. Lett. 2016, 12, 2319–2328. [Google Scholar] [CrossRef]
- Peculis, R.; Balcere, I.; Radovica-Spalvina, I.; Konrade, I.; Caune, O.; Megnis, K.; Rovite, V.; Stukens, J.; Nazarovs, J.; Breiksa, A.; et al. Case report: Recurrent pituitary adenoma has increased load of somatic variants. BMC Endocr. Disord. 2020, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Galland, F.; Lacroix, L.; Saulnier, P.; Dessen, P.; Meduri, G.; Bernier, M.; Gaillard, S.; Guibourdenche, J.; Fournier, T.; Evain-Brion, D.; et al. Differential gene expression profiles of invasive and non-invasive non-functioning pituitary adenomas based on microarray analysis. Endocr. Relat. Cancer 2010, 17, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, L.J.T.; Lerario, A.M.; de Castro, M.; Martins, C.S.; Bronstein, M.D.; Machado, M.C.; Trarbach, E.B.; Villares Fragoso, M.C.B. Transcriptome Analysis Showed a Differential Signature between Invasive and Non-invasive Corticotrophinomas. Front. Endocrinol. (Lausanne) 2017, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lekva, T.; Berg, J.P.; Fougner, S.L.; Olstad, O.K.; Ueland, T.; Bollerslev, J. Gene expression profiling identifies ESRP1 as a potential regulator of epithelial mesenchymal transition in somatotroph adenomas from a large cohort of patients with acromegaly. J. Clin. Endocrinol. Metab. 2012, 97, E1506–E1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Jian, F.; Jiang, H.; Sun, Y.; Pan, S.; Gu, C.; Chen, X.; Wang, W.; Ning, G.; Bian, L.; et al. Decreased expression of SFRP2 promotes development of the pituitary corticotroph adenoma by upregulating Wnt signaling. Int. J. Oncol. 2018, 52, 1934–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, C.; Wang, J.; Song, G.; Zhao, Z.; Wang, H.; Wang, W.; Li, H.; Li, Z.; Miao, Y.; et al. Genome-wide analysis of differentially expressed lncRNAs and mRNAs in primary gonadotrophin adenomas by RNA-seq. Oncotarget 2017, 8, 4585–4606. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, K.A.; Knox, A.J.; Xu, M.; Kiseljak-Vassiliades, K.; Edwards, M.G.; Geraci, M.; Kleinschmidt-DeMasters, B.K.; Lillehei, K.O.; Wierman, M.E. Identification of growth arrest and DNA-damage-inducible gene beta (GADD45beta) as a novel tumor suppressor in pituitary gonadotrope tumors. Endocrinology 2011, 152, 3603–3613. [Google Scholar] [CrossRef] [Green Version]
- Wierinckx, A.; Auger, C.; Devauchelle, P.; Reynaud, A.; Chevallier, P.; Jan, M.; Perrin, G.; Fèvre-Montange, M.; Rey, C.; Figarella-Branger, D.; et al. A diagnostic marker set for invasion, proliferation, and aggressiveness of prolactin pituitary tumors. Endocr. Relat. Cancer 2007, 14, 887–900. [Google Scholar] [CrossRef]
- Morris, D.G.; Musat, M.; Czirják, S.; Hanzély, Z.; Lillington, D.M.; Korbonits, M.; Grossman, A.B. Differential gene expression in pituitary adenomas by oligonucleotide array analysis. Eur. J. Endocrinol. 2005, 153, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.O.; Young, A.N.; Brown, M.R.; Brat, D.J.; Parks, J.S.; Neish, A.S.; Oyesiku, N.M. Novel patterns of gene expression in pituitary adenomas identified by complementary deoxyribonucleic acid microarrays and quantitative reverse transcription-polymerase chain reaction. J. Clin. Endocrinol. Metab. 2001, 86, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.S.; Evans, C.-O.; Zhan, X.; Okor, M.; Desiderio, D.M.; Oyesiku, N.M. Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res. 2005, 65, 10214–10222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.-O.; Moreno, C.S.; Zhan, X.; McCabe, M.T.; Vertino, P.M.; Desiderio, D.M.; Oyesiku, N.M. Molecular pathogenesis of human prolactinomas identified by gene expression profiling, RT-qPCR, and proteomic analyses. Pituitary 2008, 11, 231–245. [Google Scholar] [CrossRef]
- Cai, T.; Xiao, J.; Wang, Z.-F.; Liu, Q.; Wu, H.; Qiu, Y.-Z. Identification of differentially coexpressed genes in gonadotrope tumors and normal pituitary using bioinformatics methods. Pathol. Oncol. Res. 2014, 20, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yin, H.; Li, B.; Yang, H. Identification of Transcriptional Metabolic Dysregulation in Subtypes of Pituitary Adenoma by Integrated Bioinformatics Analysis. Diabetes Metab. Syndr. Obes. 2019, 12, 2441–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassarino, M.F.; Ambrogio, A.G.; Cassarino, A.; Terreni, M.R.; Gentilini, D.; Sesta, A.; Cavagnini, F.; Losa, M.; Pecori Giraldi, F. Gene expression profiling in human corticotroph tumours reveals distinct, neuroendocrine profiles. J. Neuroendocrinol. 2018, 30, e12628. [Google Scholar] [CrossRef]
- Yu, S.-Y.; Hong, L.-C.; Feng, J.; Wu, Y.-T.; Zhang, Y.-Z. Integrative proteomics and transcriptomics identify novel invasive-related biomarkers of non-functioning pituitary adenomas. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 8923–8930. [Google Scholar] [CrossRef]
- Cao, C.; Wang, W.; Ma, C.; Jiang, P. Computational analysis identifies invasion-associated genes in pituitary adenomas. Mol. Med. Rep. 2015, 12, 1977–1982. [Google Scholar] [CrossRef]
- Chen, Y.; Chuan, H.-L.; Yu, S.-Y.; Li, C.-Z.; Wu, Z.-B.; Li, G.-L.; Zhang, Y.-Z. A Novel Invasive-Related Biomarker in Three Subtypes of Nonfunctioning Pituitary Adenomas. World Neurosurg. 2017, 100, 514–521. [Google Scholar] [CrossRef]
- Marko, N.F.; Coughlan, C.; Weil, R.J. Towards an integrated molecular and clinical strategy to predict early recurrence in surgically resected non-functional pituitary adenomas. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2012, 19, 1535–1540. [Google Scholar] [CrossRef]
- Hernández-Ramírez, L.C.; Morgan, R.M.L.; Barry, S.; D’Acquisto, F.; Prodromou, C.; Korbonits, M. Multi-chaperone function modulation and association with cytoskeletal proteins are key features of the function of AIP in the pituitary gland. Oncotarget 2018, 9, 9177–9198. [Google Scholar] [CrossRef] [Green Version]
- Barry, S.; Carlsen, E.; Marques, P.; Stiles, C.E.; Gadaleta, E.; Berney, D.M.; Roncaroli, F.; Chelala, C.; Solomou, A.; Herincs, M.; et al. Tumor microenvironment defines the invasive phenotype of AIP-mutation-positive pituitary tumors. Oncogene 2019, 38, 5381–5395. [Google Scholar] [CrossRef]
- Richardson, T.E.; Shen, Z.-J.; Kanchwala, M.; Xing, C.; Filatenkov, A.; Shang, P.; Barnett, S.; Abedin, Z.; Malter, J.S.; Raisanen, J.M.; et al. Aggressive Behavior in Silent Subtype III Pituitary Adenomas May Depend on Suppression of Local Immune Response: A Whole Transcriptome Analysis. J. Neuropathol. Exp. Neurol. 2017, 76, 874–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saksis, R.; Silamikelis, I.; Laksa, P.; Megnis, K.; Peculis, R.; Mandrika, I.; Rogoza, O.; Petrovska, R.; Balcere, I.; Konrade, I.; et al. Medication for Acromegaly Reduces Expression of MUC16, MACC1 and GRHL2 in Pituitary Neuroendocrine Tumour Tissue. Front. Oncol. 2021, 10, 3224. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.T.; Vesely, M.D.; Miyagishima, D.F. In silico analysis of the immunological landscape of pituitary adenomas. J. Neurooncol. 2020, 147, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, C.; Zhang, D.; Peng, J.; Ma, S.; Wang, X.; Guan, X.; Li, P.; Li, D.; Jia, G.; et al. Comprehensive analysis of the immunological landscape of pituitary adenomas: Implications of immunotherapy for pituitary adenomas. J. Neurooncol. 2020, 149, 473–487. [Google Scholar] [CrossRef]
- Marques, P.; Barry, S.; Carlsen, E.; Collier, D.; Ronaldson, A.; Awad, S.; Dorward, N.; Grieve, J.; Mendoza, N.; Muquit, S.; et al. Chemokines modulate the tumour microenvironment in pituitary neuroendocrine tumours. Acta Neuropathol. Commun. 2019, 7, 172. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Lu, M.; Cheng, T.; Zhan, X.; Zhan, X. Multiomics-Based Signaling Pathway Network Alterations in Human Non-functional Pituitary Adenomas. Front. Endocrinol. (Lausanne) 2019, 10, 835. [Google Scholar] [CrossRef]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database (Oxford) 2010, 2010, baq020. [Google Scholar] [CrossRef]
- Marques, P.; Barry, S.; Carlsen, E.; Collier, D.; Ronaldson, A.; Awad, S.; Dorward, N.; Grieve, J.; Mendoza, N.; Muquit, S.; et al. Pituitary tumour fibroblast-derived cytokines influence tumour aggressiveness. Endocr. Relat. Cancer 2019, 26, 853–865. [Google Scholar] [CrossRef]
- Barbieri, F.; Bajetto, A.; Stumm, R.; Pattarozzi, A.; Porcile, C.; Zona, G.; Dorcaratto, A.; Ravetti, J.-L.; Minuto, F.; Spaziante, R.; et al. Overexpression of stromal cell-derived factor 1 and its receptor CXCR4 induces autocrine/paracrine cell proliferation in human pituitary adenomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5022–5032. [Google Scholar] [CrossRef] [Green Version]
- Ilie, M.D.; Vasiljevic, A.; Raverot, G.; Bertolino, P. The Microenvironment of Pituitary Tumors-Biological and Therapeutic Implications. Cancers 2019, 11, 1605. [Google Scholar] [CrossRef] [Green Version]
- Marques, P.; Grossman, A.B.; Korbonits, M. The tumour microenvironment of pituitary neuroendocrine tumours. Front. Neuroendocrinol. 2020, 58, 100852. [Google Scholar] [CrossRef]
- Barbieri, F.; Thellung, S.; Würth, R.; Gatto, F.; Corsaro, A.; Villa, V.; Nizzari, M.; Albertelli, M.; Ferone, D.; Florio, T. Emerging Targets in Pituitary Adenomas: Role of the CXCL12/CXCR4-R7 System. Int. J. Endocrinol. 2014, 2014, 753524. [Google Scholar] [CrossRef] [PubMed]
- Arteaga-Vázquez, M.; Caballero-Pérez, J.; Vielle-Calzada, J.-P. A family of microRNAs present in plants and animals. Plant Cell 2006, 18, 3355–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starega-Roslan, J.; Krol, J.; Koscianska, E.; Kozlowski, P.; Szlachcic, W.J.; Sobczak, K.; Krzyzosiak, W.J. Structural basis of microRNA length variety. Nucleic Acids Res. 2011, 39, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, D.P.; Kay, E.W.; Leader, M.; Murphy, G.M.; Atkins, G.J.; Mabruk, M.J. A high degree of chromosomal instability at 13q14 in cutaneous squamous cell carcinomas: Indication for a role of a tumour suppressor gene other than Rb. Mol. Pathol. 2001, 54, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Bottoni, A.; Piccin, D.; Tagliati, F.; Luchin, A.; Zatelli, M.C.; degli Uberti, E.C. miR-15a and miR-16-1 down-regulation in pituitary adenomas. J. Cell. Physiol. 2005, 204, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Tung, S.-L.; Huang, W.-C.; Hsu, F.-C.; Yang, Z.-P.; Jang, T.-H.; Chang, J.-W.; Chuang, C.-M.; Lai, C.-R.; Wang, L.-H. miRNA-34c-5p inhibits amphiregulin-induced ovarian cancer stemness and drug resistance via downregulation of the AREG-EGFR-ERK pathway. Oncogenesis 2017, 6, e326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-A.; Lee, C.-T.; Lee, J.-C.; Wang, Y.-W.; Huang, C.-T.; Lan, S.-H.; Lin, P.-C.; Lin, B.-W.; Tian, Y.-F.; Liu, H.-S.; et al. MiR-338-5p promotes metastasis of colorectal cancer by inhibition of phosphatidylinositol 3-kinase, catalytic subunit type 3-mediated autophagy pathway. EBioMedicine 2019, 43, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, J.; Wei, W.; Qi, X.; Li, C.; Ren, J. The dual-inhibitory effect of miR-338-5p on the multidrug resistance and cell growth of hepatocellular carcinoma. Signal Transduct. Target. Ther. 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Li, X. Molecular Network Basis of Invasive Pituitary Adenoma: A Review. Front. Endocrinol. (Lausanne) 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Cao, Y.; Su, T.; Jiang, Y.; Jiang, L.; Zhou, W.; Zhang, C.; Wang, W.; Ning, G. Downregulation of miR-375 in aldosterone-producing adenomas promotes tumour cell growth via MTDH. Clin. Endocrinol. (Oxf.) 2015, 83, 581–589. [Google Scholar] [CrossRef]
- Zhang, C.; Qian, Y.; Qiao, Y.; Li, Y.; Wang, W.; Li, J.; Deng, X. Analysis of whole genome-wide microRNA transcriptome profiling in invasive pituitary adenomas and non-invasive pituitary adenomas. Chin. Neurosurg. J. 2019, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yang, C.; Yang, S.; Cheng, F.; Rao, J.; Wang, X. miR-665 promotes hepatocellular carcinoma cell migration, invasion, and proliferation by decreasing Hippo signaling through targeting PTPRB. Cell Death Dis. 2018, 9, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicchio, T.M.; Aliquò, F.; Ruggeri, R.M.; Ragonese, M.; Giuffrida, G.; Cotta, O.R.; Spagnolo, F.; Torre, M.L.; Alibrandi, A.; Asmundo, A.; et al. MicroRNAs expression in pituitary tumors: Differences related to functional status, pathological features, and clinical behavior. J. Endocrinol. Investig. 2020, 43, 947–958. [Google Scholar] [CrossRef]
- Yao, J.; Wu, X. Upregulation Of miR-149-3p Suppresses Spinal Chordoma Malignancy by Targeting Smad3. Onco Targets Ther. 2019, 12, 9987–9997. [Google Scholar] [CrossRef] [Green Version]
- Zhenye, L.; Chuzhong, L.; Youtu, W.; Xiaolei, L.; Lei, C.; Lichuan, H.; Hongyun, W.; Yonggang, W.; Fei, W.; Yazhuo, Z. The expression of TGF-β1, Smad3, phospho-Smad3 and Smad7 is correlated with the development and invasion of nonfunctioning pituitary adenomas. J. Transl. Med. 2014, 12, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souteiro, P.; Karavitaki, N. Dopamine agonist resistant prolactinomas: Any alternative medical treatment? Pituitary 2020, 23, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.B.; Li, W.Q.; Lin, S.J.; De Wang, C.; Cai, L.; Lu, J.L.; Chen, Y.X.; Su, Z.P.; Shang, H.B.; Yang, W.L.; et al. MicroRNA expression profile of bromocriptine-resistant prolactinomas. Mol. Cell. Endocrinol. 2014, 395, 10–18. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, Y.; Chen, J.; Hu, Y.; Cao, Z.; Ren, P.; Zhang, Y. p21 overexpression sensitizes osteosarcoma U2OS cells to cisplatin via evoking caspase-3 and Bax/Bcl-2 cascade. Tumour Biol. J. Int. Soc. Oncodevelopmental. Biol. Med. 2014, 35, 3119–3123. [Google Scholar] [CrossRef]
- Ziller, C.; Lincet, H.; Muller, C.D.; Staedel, C.; Behr, J.-P.; Poulain, L. The cyclin-dependent kinase inhibitor p21(cip1/waf1) enhances the cytotoxicity of ganciclovir in HSV-tk transfected ovarian carcinoma cells. Cancer Lett. 2004, 212, 43–52. [Google Scholar] [CrossRef]
- Wu, Z.; Cai, L.; Lu, J.; De Wang, C.; Guan, J.; Chen, X.; Wu, J.; Zheng, W.; Wu, Z.; Li, Q.; et al. MicroRNA-93 mediates cabergoline-resistance by targeting ATG7 in prolactinoma. J. Endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Leng, Z.G.; Guo, Y.H.; Cai, L.; Cai, Y.; Li, N.; Shang, H.B.; Le, W.-D.; Zhao, W.G.; Wu, Z.B. Suppression of mTOR pathway and induction of autophagy-dependent cell death by cabergoline. Oncotarget 2015, 6, 39329–39341. [Google Scholar] [CrossRef] [Green Version]
- Izzotti, A.; Carozzo, S.; Pulliero, A.; Zhabayeva, D.; Ravetti, J.L.; Bersimbaev, R. Extracellular MicroRNA in liquid biopsy: Applicability in cancer diagnosis and prevention. Am. J. Cancer Res. 2016, 6, 1461–1493. [Google Scholar]
- Wang, Q.; Li, P.; Li, A.; Jiang, W.; Wang, H.; Wang, J.; Xie, K. Plasma specific miRNAs as predictive biomarkers for diagnosis and prognosis of glioma. J. Exp. Clin. Cancer Res. 2012, 31, 97. [Google Scholar] [CrossRef] [Green Version]
- Németh, K.; Darvasi, O.; Likó, I.; Szücs, N.; Czirják, S.; Reiniger, L.; Szabó, B.; Krokker, L.; Pállinger, É.; Igaz, P.; et al. Comprehensive analysis of circulating microRNAs in plasma of patients with pituitary adenomas. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.T.; Nieman, L.K.; Feelders, R.A. Cushing’s syndrome: Epidemiology and developments in disease management. Clin. Epidemiol. 2015, 7, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarial, K.D.S.; Bhansali, A.; Gupta, V.; Singh, P.; Mukherjee, K.K.; Sharma, A.; Vashishtha, R.K.; Sukumar, S.P.; Sachdeva, N.; Walia, R. Diagnostic accuracy and comparison of BIPSS in response to lysine vasopressin and hCRH. Endocr. Connect. 2018, 7, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaya, Z.; Khandaeva, P.; Nonn, L.; Nikitin, A.; Solodovnikov, A.; Sitkin, I.; Grigoriev, A.; Pikunov, M.; Lapshina, A.; Rozhinskaya, L.; et al. Circulating Plasma microRNA to Differentiate Cushing’s Disease From Ectopic ACTH Syndrome. Front. Endocrinol. (Lausanne) 2020, 11, 331. [Google Scholar] [CrossRef]
- Wu, X.-L.; Cheng, B.; Li, P.-Y.; Huang, H.-J.; Zhao, Q.; Dan, Z.-L.; Tian, D.-A.; Zhang, P. MicroRNA-143 suppresses gastric cancer cell growth and induces apoptosis by targeting COX-2. World J. Gastroenterol. 2013, 19, 7758–7765. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Dai, G.; Yu, L.; Hu, Q.; Chen, J.; Guo, W. miR-143-3p inhibits the proliferation, migration and invasion in osteosarcoma by targeting FOSL2. Sci. Rep. 2018, 8, 606. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ma, D.; Liu, H.; Wang, J.; Fan, J.; Li, X. MicroRNA-143 shows tumor suppressive effects through inhibition of oncogenic K-Ras in pituitary tumor. Int. J. Clin. Exp. Pathol. 2017, 10, 10969–10978. [Google Scholar] [PubMed]
- Jian, M.; Du, Q.; Zhu, D.; Mao, Z.; Wang, X.; Feng, Y.; Xiao, Z.; Wang, H.; Zhu, Y. Tumor suppressor miR-145-5p sensitizes prolactinoma to bromocriptine by downregulating TPT1. J. Endocrinol. Investig. 2019, 42, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, H.; Tao, S.; Zheng, Y.; Wu, W.; Lian, F.; Jaramillo, M.; Fang, D.; Zhang, D.D. Tumor protein translationally controlled 1 is a p53 target gene that promotes cell survival. Cell Cycle 2013, 12, 2321–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yang, K.; Ren, T.; Huang, Y.; Tang, X.; Guo, W. miR-16-5p inhibits chordoma cell proliferation, invasion and metastasis by targeting Smad3. Cell Death Dis. 2018, 9, 680. [Google Scholar] [CrossRef]
- Qu, Y.; Liu, H.; Lv, X.; Liu, Y.; Wang, X.; Zhang, M.; Zhang, X.; Li, Y.; Lou, Q.; Li, S.; et al. MicroRNA-16-5p overexpression suppresses proliferation and invasion as well as triggers apoptosis by targeting VEGFA expression in breast carcinoma. Oncotarget 2017, 8, 72400–72410. [Google Scholar] [CrossRef]
- Gu, Z.; Li, Z.; Xu, R.; Zhu, X.; Hu, R.; Xue, Y.; Xu, W. miR-16-5p Suppresses Progression and Invasion of Osteosarcoma via Targeting at Smad3. Front. Pharmacol. 2020, 11, 1324. [Google Scholar] [CrossRef]
- Ruan, L.; Qian, X. MiR-16-5p inhibits breast cancer by reducing AKT3 to restrain NF-κB pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonte, F.; Santamaria, G.; Sacco, A.; Perrone, F.M.; Di Cello, A.; Battaglia, A.M.; Salatino, A.; Di Vito, A.; Aversa, I.; Venturella, R.; et al. MicroRNA let-7g acts as tumor suppressor and predictive biomarker for chemoresistance in human epithelial ovarian cancer. Sci. Rep. 2019, 9, 5668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Zhao, X.; Jin, Z.; Hou, M. Hsa-let-7g miRNA regulates the anti-tumor effects of gastric cancer cells under oxidative stress through the expression of DDR genes. J. Toxicol. Sci. 2015, 40, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han Li, C.; Chen, Y. Small and Long Non-Coding RNAs: Novel Targets in Perspective Cancer Therapy. Curr. Genom. 2015, 16, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA-LncRNA Interactions. Methods Mol. Biol. 2016, 1402, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [Green Version]
- Gejman, R.; Batista, D.L.; Zhong, Y.; Zhou, Y.; Zhang, X.; Swearingen, B.; Stratakis, C.A.; Hedley-Whyte, E.T.; Klibanski, A. Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab. 2008, 93, 4119–4125. [Google Scholar] [CrossRef]
- Tian, M.; Gong, W.; Guo, J. Long non-coding RNA SNHG1 indicates poor prognosis and facilitates disease progression in acute myeloid leukemia. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lu, Q.; Zhu, D.; Han, Y.; Zhou, X.; Ren, T. Lnc-SNHG1 may promote the progression of non-small cell lung cancer by acting as a sponge of miR-497. Biochem. Biophys. Res. Commun. 2018, 506, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, G.; Gao, Y.; Zhao, C.; Li, X.; Zhang, F.; Jiang, C.; Wu, B. Lnc-SNHG1 Activates the TGFBR2/SMAD3 and RAB11A/Wnt/β-Catenin Pathway by Sponging MiR-302/372/373/520 in Invasive Pituitary Tumors. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Y.T.; Tang, H.; Xie, W.Q.; Yao, H.; Gu, W.T.; Zheng, Y.Z.; Shang, H.B.; Wang, Y.; Wei, Y.X.; et al. Exosome-Transmitted lncRNA H19 Inhibits the Growth of Pituitary Adenoma. J. Clin. Endocrinol. Metab. 2019, 104, 6345–6356. [Google Scholar] [CrossRef] [PubMed]
- Megnis, K.; Peculis, R.; Rovite, V.; Laksa, P.; Niedra, H.; Balcere, I.; Caune, O.; Breiksa, A.; Nazarovs, J.; Stukens, J.; et al. Evaluation of the Possibility to Detect Circulating Tumor DNA From Pituitary Adenoma. Front. Endocrinol. (Lausanne) 2019, 10, 615. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Publications and study design | Kim et al. [24] 3 noninvasive non-functioning vs. 11 invasive non-functioning PitNET | Falch et al., 2018 [27] 4 fast growing vs. 4 slow growing gonadotroph PitNET | Cao et al., 2015 [98] 8 invasive vs. 8 non-invasive PitNET | Wierinckx et al., 2007 [89] 3 non-invasive vs. 4 invasive vs. 3 aggressive prolactinomas | Chen et al., 2017 [99] 3 invasive non-functioning vs. 4 invasive non-functioning PitNET | Yu et al., 2016 [97] 3 invasive non-functioning vs. 4 invasive non-functioning PitNET | De Araujo et al., 2017 [84] 4 non-invasive micro vs. 5 non-invasive macro vs. 3 invasive macro corticotroph PitNET | Galland et al., 2010 [83] 22 invasive vs. 18 non-invasive gonadotroph PitNET |
| Falch et al., 2018 [27] 4 fast growing vs. 4 slow growing gonadotroph PitNET | SERPING1; AEBP1 | |||||||
| Cao et al., 2015 [98] 8 invasive vs. 8 non-invasive PitNET | FRMPD1 | F3 | ||||||
| Wierinckx et al., 2007 [89] 3 non-invasive vs. 4 invasive vs. 3 aggressive prolactinomas | no overlap | TRIM36 1 | GAL3ST3 | |||||
| Chen et al., 2017 [99] 3 invasive vs. 4 invasive non-functioning PitNET | no overlap | no overlap | no overlap | no overlap | ||||
| Yu et al., 2016 [97] 3 invasive vs. 4 invasive non-functioning PitNET | no overlap | no overlap | POR | no overlap | EZR; SLC2A1 | |||
| De Araujo et al., 2017 [84] 4 non-invasive micro vs. 5 non-invasive macro vs. 3 invasive macro corticotroph PitNET | TGM2 | SLC1A2; RYR2; ZNF676 | no overlap | no overlap | no overlap | no overlap | ||
| Galland et al., 2010 [83] 22 invasive vs. 18 non-invasive gonadotroph PitNET | no overlap | no overlap | no overlap | no overlap | no overlap | no overlap | CD200 | |
| Levka et al., 2012 [85] 8 low cadherin vs. 8 high cadherin somatotroph PitNET | SLC24A2; FAT1 | DCLK1; TRIM36 | no overlap | TRIM36 | no overlap | no overlap | VAT1L; ELAVL3; CDKN1B; SEZ6L; NIN; FXYD5; SEPT3; CCND2; SV2B | no overlap |
| Publications and study design | Ren et al., 2018 [86] PitNET vs. normal pituitary tissue (ACTH secreting) | Evans et al., 2008 [93] PitNET vs. normal pituitary tissue (PRL secreting) | Li et al., 2017 [87] PitNET vs. normal pituitary tissue (gonadotroph secreting) | Cai et al., 2014 [94] PitNET vs. normal pituitary tissue (gonadotroph secreting) | Michaelis et al., 2011 [88] PitNET vs. normal pituitary tissue (gonadotroph secreting) | Moreno et al., 2005 [92] PitNET vs. normal pituitary tissue (nonfunctional) |
| Evans et al., 2008 [93] PitNET vs. normal pituitary tissue (PRL secreting) | not comparable 1 | |||||
| Li et al., 2017 [87] PitNET vs. normal pituitary tissue (gonadotroph secreting) | not comparable | not comparable | ||||
| Cai et al., 2014 [94] PitNET vs. normal pituitary tissue (gonadotroph secreting) | not comparable | not comparable | no overlap | |||
| Michaelis et al., 2011 [88] PitNET vs. normal pituitary tissue (gonadotroph secreting) | not comparable | not comparable | POMC2; GH1; GH2; PRL; BTG2; GAL; | F3; POR; BCAT1; COL18A1; | ||
| Moreno et al., 2005 [92] PitNET vs. normal pituitary tissue (nonfunctional) | not comparable | not comparable | not comparable | not comparable | not comparable | |
| Hu et al., 2019 [95] Different types * of PitNETs vs. normal pituitary tissue * for comparison markers identified in subgroups (ACTH secreting, PRL secreting, nonfunctional) were extracted from supplementary data separately and comparison carried out only type specifically | PMAIP1; TSHB | POMC; GH1; DLK1; GH2; PON3; CSHL1; NNAT; RBP4; IGFBP5; GPC3; GAL; NPTX2; GADD45G; CXCR4; F3; TGFBR3; GADD45B; IGFBP3; RPGR; CEBPD; GJA1; CCL2; MAFF; CDKN2A; CDKN1C; SDC4; CLDN3; POU1F1; RRAS2; | not comparable | not comparable | not comparable | GLCE; EPHB6; CABP1; DCX; EFNB3; CSPG5; PITX2; GNB3; GATA3; PBX3; HIST2H2BE; SPOCK3; NLGN1; IDH1; ATP1B2; ENO2; DPYSL3; KCNK3; VSNL1; FAIM2; SLC22A4; TM7SF2; SEZ6L; EPS8; FOLR1; IFI44; STC1; KCNJ6; ODC1; DUSP4; COL4A5; KDELR3; BBOX1; NPTX2; NNAT; CREM; HTATSF1; ID3; AMOT; THBS2; NR4A2; AGR2; RGS16; CEL; CGA; PMAIP1; KLK11; ID4; IMPA2; ID1; CCL2; CSHL1; BLM; PON3; PRL; GAL; SELL; THBS4; DLK1; GH2; GH1; |
| miRNA | Expression | Sample Type | PitNET Type | Reference Sample Set | Study |
|---|---|---|---|---|---|
| miR-15a | 🡫 | Tumor tissue | GH, PRL | Normal pituitary tissues | Bottoni et al., 2005 [120] |
| miR-16-1 | 🡫 | Tumor tissue | GH, PRL | Normal pituitary tissues | Bottoni et al., 2005 [120] |
| miR-34c-5p | 🡫 | Tumor tissue | PRL | Normal pituitary tissues | He et al., 2019 [30] |
| miR-338-5p | 🡫 | Tumor tissue | PRL | Normal pituitary tissues | He et al., 2019 [30] |
| miR-378 | 🡫 | Tumor tissue | PRL | Normal pituitary tissues | He et al., 2019 [30] |
| miR-665 | 🡩 | Tumor tissue | NFPA (invasive phenotype) | NFPA (non-invasive phenotype) | Zhang et al., 2019 [122] |
| miR-149-3p | 🡫 | Tumor tissue | NFPA (invasive phenotype) | NFPA (non-invasive phenotype) | Zhang et al., 2019 [122] |
| miR-93 | 🡩 | Tumor tissue | PRL (bromocriptine resistant phenotype) | PRL (bromocriptine sensitive phenotype) | Wu et al., 2014 [133] |
| miR-17 | 🡩 | Tumor tissue | PRL (bromocriptine resistant phenotype) | PRL (bromocriptine sensitive phenotype) | Wu et al., 2014 [133] |
| miR-143-3p | 🡫 | Plasma (after resection of PitNET) | FSH/LH | Plasma (before resection of PitNET) | Németh et al., 2019 [140] |
| miR-16-5p | 🡩 | Plasma | ACTH | Plasma of ectopic CS patients | Belaya et al., 2020 [143] |
| miR-145-5p | 🡩 | Plasma | ACTH | Plasma of ectopic CS patients | Belaya et al., 2020 [143] |
| let-7g-5p | 🡩 | Plasma | ACTH | Plasma of ectopic CS patients | Belaya et al., 2020 [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peculis, R.; Niedra, H.; Rovite, V. Large Scale Molecular Studies of Pituitary Neuroendocrine Tumors: Novel Markers, Mechanisms and Translational Perspectives. Cancers 2021, 13, 1395. https://doi.org/10.3390/cancers13061395
Peculis R, Niedra H, Rovite V. Large Scale Molecular Studies of Pituitary Neuroendocrine Tumors: Novel Markers, Mechanisms and Translational Perspectives. Cancers. 2021; 13(6):1395. https://doi.org/10.3390/cancers13061395
Chicago/Turabian StylePeculis, Raitis, Helvijs Niedra, and Vita Rovite. 2021. "Large Scale Molecular Studies of Pituitary Neuroendocrine Tumors: Novel Markers, Mechanisms and Translational Perspectives" Cancers 13, no. 6: 1395. https://doi.org/10.3390/cancers13061395