
Ca2+ Signaling as the Untact Mode during Signaling in Metastatic Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Ca2+ Signaling-Associated Molecules
1.2. Types of Ca2+ Channels
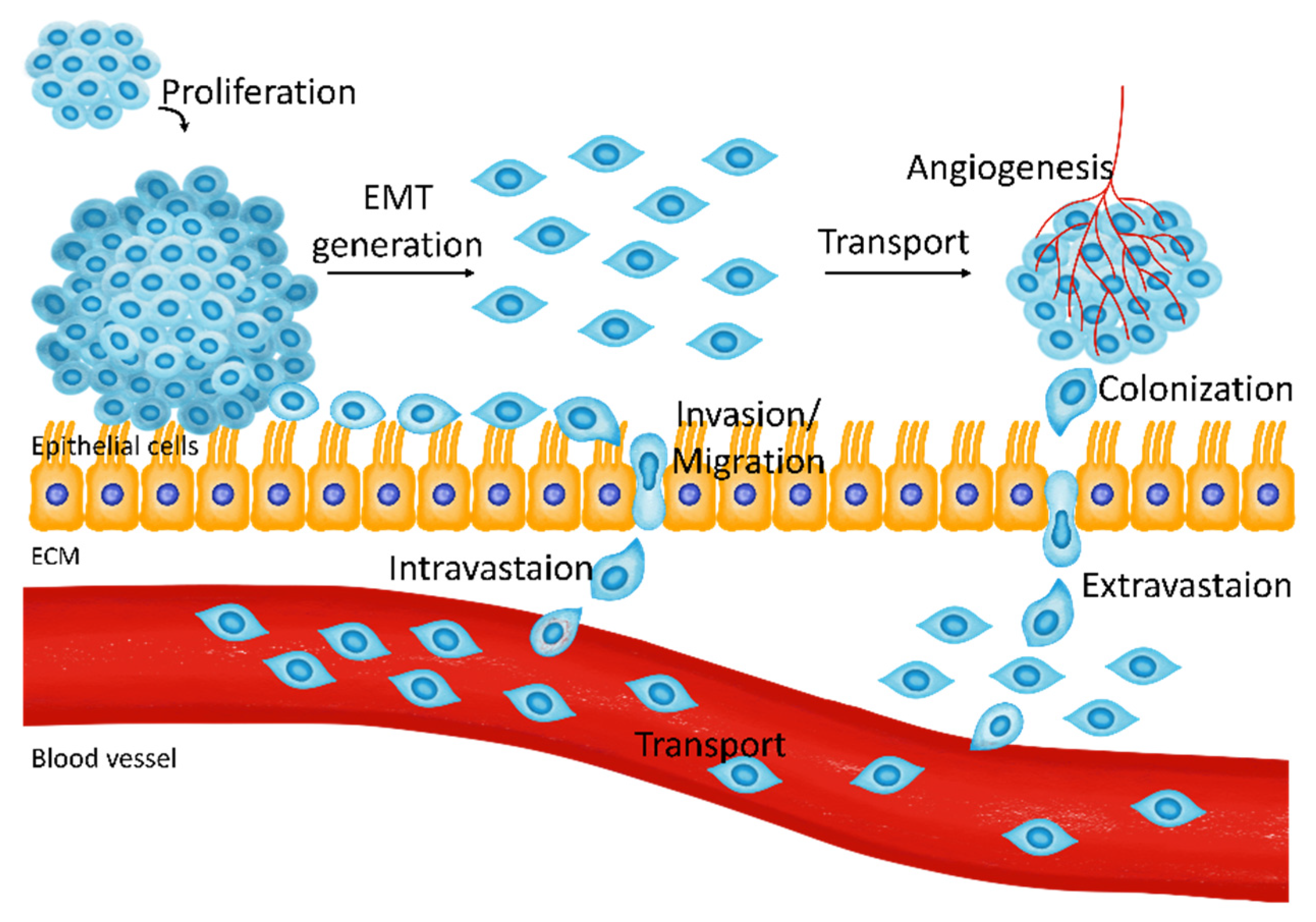
2. The Relationship between Breast Cancer Metastasis and Ca2+ Channels
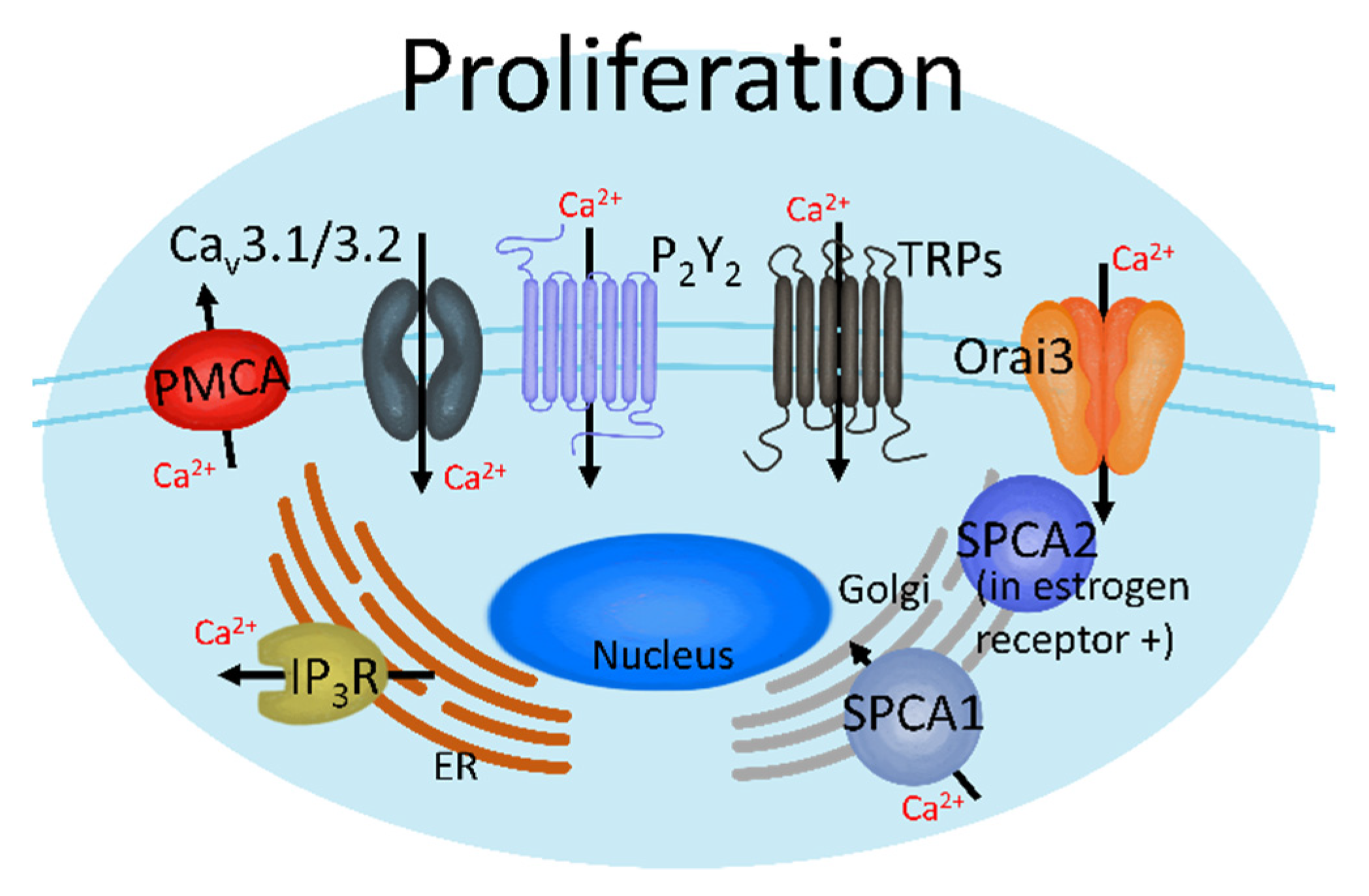
2.1. Proliferation in the Initial Metastatic Stage
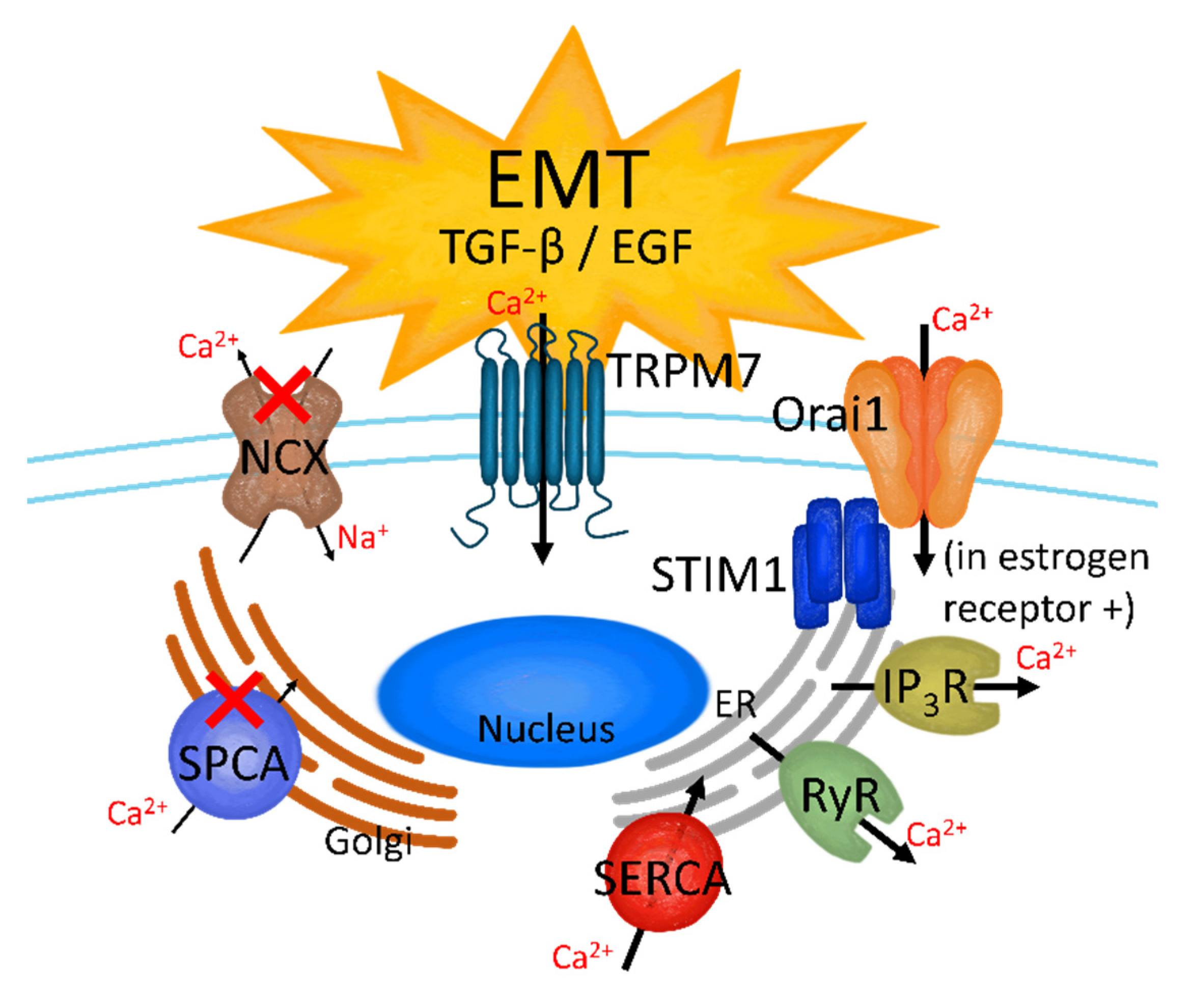
2.2. EMT
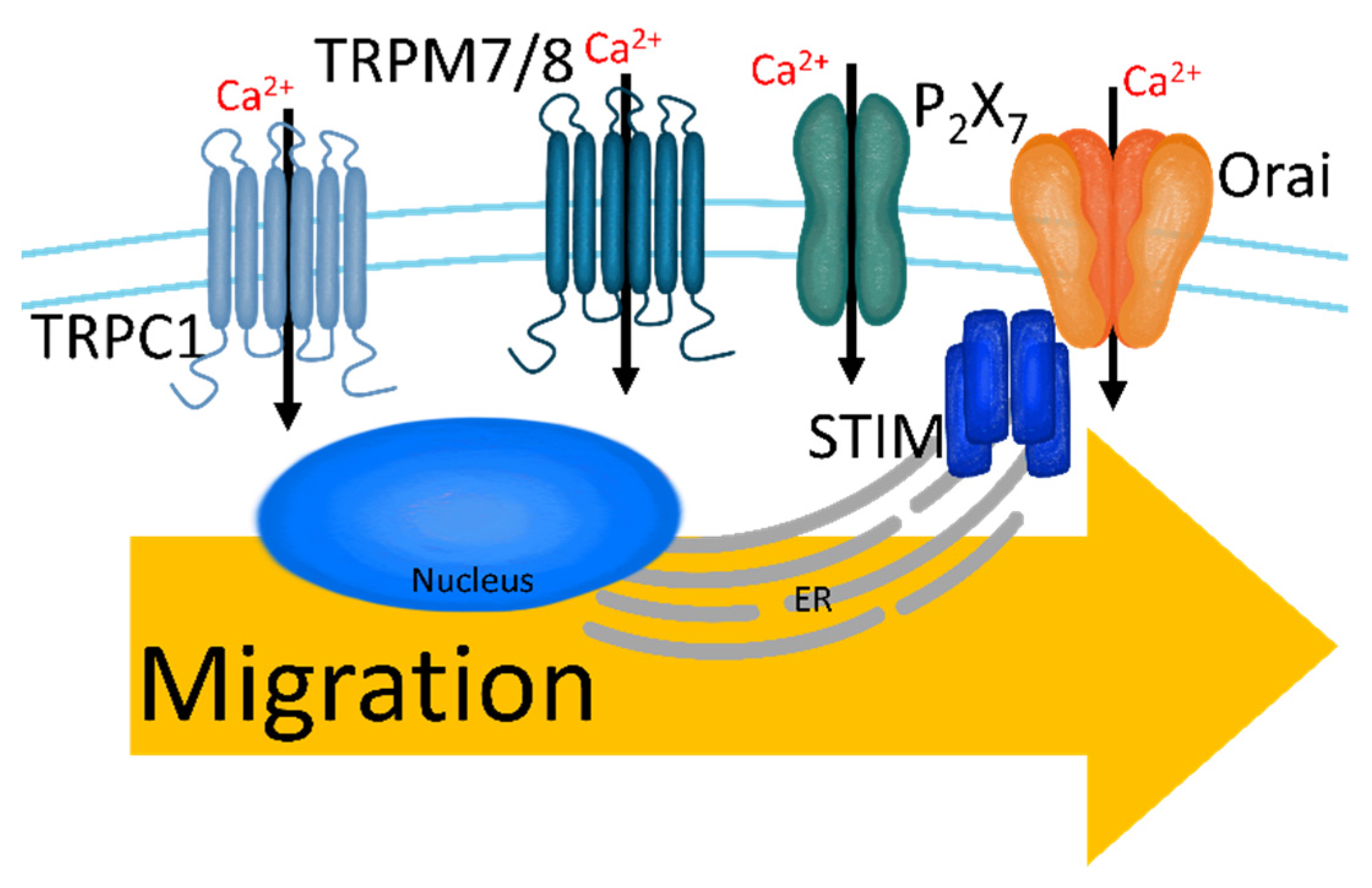
2.3. Migration and Intravasation
2.4. Colonization and Angiogenesis
3. Metastasis of Breast Tumor to the Brain
4. The Pharmacological Application of Ca2+ Signaling Blockers to Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents | Description | Effect | Ref. |
|---|---|---|---|
| Amlodipine | Medication for high blood pressure and L-type Ca2+ channel inhibitor | Decrease of HT39-transplanted breast cancer growth | [153] |
| Diltiazem | |||
| Verapamil | |||
| Mibefradil | Hypertension drug | Decrease of MCF-7 growth through inhibition of T-type Ca2+ current | [154] |
| Pimozide | Chronic psychosis drug | [155] | |
| 2-APB | TRPM7 inhibitor | Decrease of MDA-MB-231, AU565, and T47D cell growth through pausing cell cycle | [158] |
| Clotrimazole | TRPM2 inhibitor | Decrease of MDA-MB-231 cell growth through G1-phase arrest | [160] |
| Carboxyamidotriazole | Reduce mitochondrial membrane potential | Attenuation of ROS | [162] |
| Thapsigargin | SERCA inhibitor | Inhibition of S100A4 expression in MDA-MB-231 | [163] |
5. Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ksiazkiewicz, M.; Markiewicz, A.; Zaczek, A.J. Epithelial-mesenchymal transition: A hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology 2012, 79, 195–208. [Google Scholar] [CrossRef]
- Skovierova, H.; Okajcekova, T.; Strnadel, J.; Vidomanova, E.; Halasova, E. Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review). Int. J. Mol. Med. 2018, 41, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockhorn, M.; Jain, R.K.; Munn, L.L. Active versus passive mechanisms in metastasis: Do cancer cells crawl into vessels, or are they pushed? Lancet Oncol. 2007, 8, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Dua, R.S.; Gui, G.P.H.; Isacke, C.M. Endothelial adhesion molecules in breast cancer invasion into the vascular and lymphatic systems. Ejso-Eur. J. Surg. Oncol. 2005, 31, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J. The organ microenvironment and cancer metastasis. Differentiation 2002, 70, 498–505. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Neophytou, C.; Boutsikos, P.; Papageorgis, P. Molecular Mechanisms and Emerging Therapeutic Targets of Triple-Negative Breast Cancer Metastasis. Front. Oncol. 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Rubens, R.D. Metastatic breast cancer. Curr. Opin. Oncol. 1993, 5, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Rojas, K.; Stuckey, A. Breast Cancer Epidemiology and Risk Factors. Clin. Obstet. Gynecol. 2016, 59, 651–672. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Seke Etet, P.F.; Vecchio, L.; Muller, J.M.; Krampera, M.; Lukong, K.E. Signaling pathways in breast cancer: Therapeutic targeting of the microenvironment. Cell. Signal 2014, 26, 2843–2856. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Shin, H.Y.; Kim, S.H.; Lee, Y.J.; Kim, D.K. The effect of mechanical ventilation tidal volume during pneumoperitoneum on shoulder pain after a laparoscopic appendectomy. Surg. Endosc. 2010, 24, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.S.; Kim, M.S.; Park, H.S.; Lee, J.S.; Lee, J.S.; Kim, S.I.; Park, B.W. Characteristics and outcomes according to molecular subtypes of breast cancer as classified by a panel of four biomarkers using immunohistochemistry. Breast 2012, 21, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Baeg, S.; Park, I.; Kim, J.; Park, C.; Cho, H.; Yang, K.; Kim, J.; Shin, Y.; Park, K.; Gwak, G. Comparative Study for Clinical Outcomes of Triple-Positive and Triple-Negative Breast Cancer: Long-term Results in 161 Patients Followed in a Single Center. J. Breast Dis. 2020, 8, 78–84. [Google Scholar] [CrossRef]
- Eroles, P.; Bosch, A.; Perez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, A.G. Calcium regulation of muscle contraction. Biophys. J. 1975, 15, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Vig, M.; Kinet, J.P. Calcium signaling in immune cells. Nat. Immunol. 2009, 10, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambudkar, I.S. Calcium signalling in salivary gland physiology and dysfunction. J. Physiol. 2016, 594, 2813–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar] [CrossRef] [PubMed]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Fedorenko, O.A.; Popugaeva, E.; Enomoto, M.; Stathopulos, P.B.; Ikura, M.; Bezprozvanny, I. Intracellular calcium channels: Inositol-1,4,5-trisphosphate receptors. Eur. J. Pharmacol. 2014, 739, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Amador, F.J.; Stathopulos, P.B.; Enomoto, M.; Ikura, M. Ryanodine receptor calcium release channels: Lessons from structure-function studies. FEBS J. 2013, 280, 5456–5470. [Google Scholar] [CrossRef]
- Van Petegem, F. Ryanodine receptors: Allosteric ion channel giants. J. Mol. Biol. 2015, 427, 31–53. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [Green Version]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 2002, 32, 279–305. [Google Scholar] [CrossRef]
- Strehler, E.E.; Treiman, M. Calcium pumps of plasma membrane and cell interior. Curr. Mol. Med. 2004, 4, 323–335. [Google Scholar] [CrossRef]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [CrossRef]
- Lee, H.C.; Aarhus, R.; Graeff, R.; Gurnack, M.E.; Walseth, T.F. Cyclic Adp Ribose Activation of the Ryanodine Receptor Is Mediated by Calmodulin. Nature 1994, 370, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.; Hori, T.; Saitoh, N.; Takahashi, T. 4-Chloro-m-cresol, an activator of ryanodine receptors, inhibits voltage-gated K+ channels at the rat calyx of Held. Eur. J. Neurosci. 2007, 26, 1530–1536. [Google Scholar] [CrossRef]
- Hill, A.P.; Kingston, O.; Sitsapesan, R. Functional regulation of the cardiac ryanodine receptor by suramin and calmodulin involves multiple binding sites. Mol. Pharmacol. 2004, 65, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Khananshvili, D. The SLC8 gene family of sodium-calcium exchangers (NCX)—Structure, function, and regulation in health and disease. Mol. Aspects Med. 2013, 34, 220–235. [Google Scholar] [CrossRef]
- McBain, C.J.; Mayer, M.L. N-methyl-D-aspartic acid receptor structure and function. Physiol. Rev. 1994, 74, 723–760. [Google Scholar] [CrossRef] [PubMed]
- Jonas, P.; Burnashev, N. Molecular Mechanisms Controlling Calcium-Entry through Ampa-Type Glutamate-Receptor Channels. Neuron 1995, 15, 987–990. [Google Scholar] [CrossRef] [Green Version]
- Reyes, E.P.; Cerpa, V.; Corvalan, L.; Retamal, M.A. Cxs and Panx- hemichannels in peripheral and central chemosensing in mammals. Front. Cell. Neurosci. 2014, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Xie, X. Tools for GPCR drug discovery. Acta Pharmacol. Sin. 2012, 33, 372–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhyani, V.; Gare, S.; Gupta, R.K.; Swain, S.; Venkatesh, K.V.; Giri, L. GPCR mediated control of calcium dynamics: A systems perspective. Cell. Signal 2020, 74, 109717. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Miyano, K.; Sudo, Y.; Yokoyama, A.; Hisaoka-Nakashima, K.; Morioka, N.; Takebayashi, M.; Nakata, Y.; Higami, Y.; Uezono, Y. History of the G protein-coupled receptor (GPCR) assays from traditional to a state-of-the-art biosensor assay. J. Pharmacol. Sci. 2014, 126, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E.; Runnels, L.W.; Strubing, C. The TRP ion channel family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Schwab, A. Ion channels and transporters in metastasis. Biochim. Biophys. Acta 2015, 1848, 2638–2646. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjo, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Dingsdale, H.; Okeke, E.; Awais, M.; Haynes, L.; Criddle, D.N.; Sutton, R.; Tepikin, A.V. Saltatory formation, sliding and dissolution of ER-PM junctions in migrating cancer cells. Biochem. J. 2013, 451, 25–32. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Turner, K.L.; Sontheimer, H. Calcium entry via TRPC1 channels activates chloride currents in human glioma cells. Cell Calcium 2013, 53, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitru, A.; Toader, D.O.; Cretoiu, S.M.; Cretoiu, D.; Suciu, N.; Radu, B.M. Alterations in Calcium Signaling Pathways in Breast Cancer. Calcium Signal Transduct. 2018, 24, 165. [Google Scholar] [CrossRef] [Green Version]
- Tungsukruthai, S.; Petpiroon, N.; Chanvorachote, P. Molecular Mechanisms of Breast Cancer Metastasis and Potential Anti-metastatic Compounds. AntiCancer Res. 2018, 38, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- So, C.L.; Saunus, J.M.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium signalling and breast cancer. Semin. Cell Dev. Biol. 2019, 94, 74–83. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Pasdar, A.; Rezaee, M.; Fazeli, M.; Soleimanpour, S.; Hassanian, S.M.; FarshchiyanYazdi, Z.; Rad, T.Y.; Ferns, G.A.; Avan, A. The current status and perspectives regarding the clinical implication of intracellular calcium in breast cancer. J. Cell. Physiol. 2018, 233, 5623–5641. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Chagtoo, M.; Tiwari, S.; George, N.; Chakravarti, B.; Khan, S.; Lakshmi, S.; Godbole, M.M. Inhibition of Inositol 1, 4, 5-Trisphosphate Receptor Induce Breast Cancer Cell Death through Deregulated Autophagy and Cellular Bioenergetics. J. Cell. Biochem. 2017, 118, 2333–2346. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Roberts-Thomson, S.J.; Holman, N.A.; May, F.J.; Lehrbach, G.M.; Monteith, G.R. Expression of plasma membrane calcium pump isoform mRNAs in breast cancer cell lines. Cell. Signal 2002, 14, 1015–1022. [Google Scholar] [CrossRef]
- Lee, W.J.; Roberts-Thomson, S.J.; Monteith, G.R. Plasma membrane calcium-ATPase 2 and 4 in human breast cancer cell lines. Biochem. Biophys. Res. Commun. 2005, 337, 779–783. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.V.; Al-Refae, K.; Wolk, G.; Bonatz, G.; Altmuller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer 2016, 8, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolanz, K.A.; Hediger, M.A.; Landowski, C.P. The role of TRPV6 in breast carcinogenesis. Mol. Cancer Ther. 2008, 7, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grice, D.M.; Vetter, I.; Faddy, H.M.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Golgi Calcium Pump Secretory Pathway Calcium ATPase 1 (SPCA1) Is a Key Regulator of Insulin-like Growth Factor Receptor (IGF1R) Processing in the Basal-like Breast Cancer Cell Line MDA-MB-231. J. Biol. Chem. 2010, 285, 37458–37466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Cross, B.M.; Hack, A.; Reinhardt, T.A.; Rao, R. SPCA2 regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation. PLoS ONE 2013, 8, e67348. [Google Scholar] [CrossRef]
- Peters, A.A.; Milevskiy, M.J.; Lee, W.C.; Curry, M.C.; Smart, C.E.; Saunus, J.M.; Reid, L.; da Silva, L.; Marcial, D.L.; Dray, E.; et al. The calcium pump plasma membrane Ca2+-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci. Rep. 2016, 6, 25505. [Google Scholar] [CrossRef]
- Curry, M.; Roberts-Thomson, S.J.; Monteith, G.R. PMCA2 silencing potentiates MDA-MB-231 breast cancer cell death initiated with the Bcl-2 inhibitor ABT-263. Biochem. Biophys. Res. Commun. 2016, 478, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.T.; Huang, L.; Pottle, J.E.; Liu, K.; Yang, Y.; Zeng, X.; Keyser, B.M.; Agrawal, K.C.; Hansen, J.B.; Li, M. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008, 267, 116–124. [Google Scholar] [CrossRef]
- Szatkowski, C.; Parys, J.B.; Ouadid-Ahidouch, H.; Matifat, F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Eun, S.Y.; Lee, J.S.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. P2Y2 receptor activation by nucleotides released from highly metastatic breast cancer cells increases tumor growth and invasion via crosstalk with endothelial cells. Breast Cancer Res. 2014, 16, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hiani, Y.; Lehen’kyi, V.; Ouadid-Ahidouch, H.; Ahidouch, A. Activation of the calcium-sensing receptor by high calcium induced breast cancer cell proliferation and TRPC1 cation channel over-expression potentially through EGFR pathways. Arch. Biochem. Biophys. 2009, 486, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Aydar, E.; Yeo, S.; Djamgoz, M.; Palmer, C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: A potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell Physiol. 2009, 297, C493–C502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, Q.; Meng, H.; Jiang, Y.; Mao, A.; Wang, T.; Hua, D.; Yao, X.; Jin, J.; Ma, X. Enhancement of vascular endothelial growth factor release in long-term drug-treated breast cancer via transient receptor potential channel 5-Ca2+-hypoxia-inducible factor 1alpha pathway. Pharmacol. Res. 2015, 93, 36–42. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yang, D.; Myeong, J.; Ha, K.; Kim, S.H.; Park, E.J.; Kim, I.G.; Cho, N.H.; Lee, K.P.; Jeon, J.H.; et al. Regulation of calcium influx and signaling pathway in cancer cells via TRPV6-Numb1 interaction. Cell Calcium 2013, 53, 102–111. [Google Scholar] [CrossRef]
- Samatov, T.R.; Tonevitsky, A.G.; Schumacher, U. Epithelial-mesenchymal transition: Focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol. Cancer 2013, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, S.H.; Cheng, H.; Li, J.; Feng, R. The effect of TGF-beta-induced epithelial-mesenchymal transition on the expression of intracellular calcium-handling proteins in T47D and MCF-7 human breast cancer cells. Arch. Biochem. Biophys. 2015, 583, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Parsonage, M.T.; Cabot, P.J.; Parat, M.O.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int. 2013, 13, 76. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Qin, K.; Zhang, Y.; Gong, J.; Li, N.; Lv, D.; Xiang, R.; Tan, X. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 411, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, T.A.; Azimi, I.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. A role for calcium in the regulation of ATP-binding cassette, sub-family C, member 3 (ABCC3) gene expression in a model of epidermal growth factor-mediated breast cancer epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2015, 458, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, D.K.; Makena, M.R.; Llongueras, J.P.; Prasad, H.; Ko, M.; Bandral, M.; Rao, R. A Ca2+-ATPase Regulates E-cadherin Biogenesis and Epithelial-Mesenchymal Transition in Breast Cancer Cells. Mol. Cancer Res. 2019, 17, 1735–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Miao, Y.; Zheng, X.; Gong, Y.; Zhang, J.; Zou, F.; Cai, C. STIM1 and STIM2 differently regulate endogenous Ca2+ entry and promote TGF-beta-induced EMT in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 488, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Schaar, A.; Sukumaran, P.; Sun, Y.; Dhasarathy, A.; Singh, B.B. TRPC1-STIM1 activation modulates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Oncotarget 2016, 7, 80554–80567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.T.D.S.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.M.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W.; Goodhill, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014, 33, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.T.; Jiang, X.H.; Xie, C.; Cheng, H.; Dong, J.D.; Wang, Y.; Fok, K.L.; Zhang, X.H.; Sun, T.T.; Tsang, L.L.; et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Bba-Mol. Cell Res. 2013, 1833, 2961–2969. [Google Scholar] [CrossRef] [Green Version]
- Iamshanova, O.; Fiorio Pla, A.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [Green Version]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef]
- Davis, F.M.; Kenny, P.A.; Soo, E.T.L.; van Denderen, B.J.W.; Thompson, E.W.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Remodeling of Purinergic Receptor-Mediated Ca2+ Signaling as a Consequence of EGF-Induced Epithelial-Mesenchymal Transition in Breast Cancer Cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Beilby, H.; Davis, F.M.; Marcial, D.L.; Kenny, P.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Altered purinergic receptor-Ca2+ signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol. Oncol. 2016, 10, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.C.; Meyer, T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 2012, 22, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betapudi, V. Life without double-headed non-muscle myosin II motor proteins. Front. Chem. 2014, 2, 45. [Google Scholar] [CrossRef] [Green Version]
- Betapudi, V.; Rai, V.; Beach, J.R.; Egelhoff, T. Novel regulation and dynamics of myosin II activation during epidermal wound responses. Exp. Cell Res. 2010, 316, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.L.; Davies, P.L. Structure-function relationships in calpains. Biochem. J. 2012, 447, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daft, P.G.; Yuan, K.; Warram, J.M.; Klein, M.J.; Siegal, G.P.; Zayzafoon, M. Alpha-CaMKII plays a critical role in determining the aggressive behavior of human osteosarcoma. Mol. Cancer Res. 2013, 11, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Ronde, P.; Gaire, M.; Haiech, J.; Takeda, K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J. Biol. Chem. 2002, 277, 26364–26371. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.C.; Chen, W.T. Cellular invasion into matrix beads: Localization of beta 1 integrins and fibronectin to the invadopodia. J. Cell Sci. 1991, 99 Pt 2, 213–225. [Google Scholar]
- Caldieri, G.; Buccione, R. Aiming for invadopodia: Organizing polarized delivery at sites of invasion. Trends Cell Biol. 2010, 20, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Lai, C.S.; Chen, Y.F.; Chiu, W.T.; Chen, H.C.; Shen, M.R. STIM1-dependent Ca2+ signaling regulates podosome formation to facilitate cancer cell invasion. Sci. Rep. 2017, 7, 11523. [Google Scholar] [CrossRef] [Green Version]
- Di, J.; Huang, H.; Qu, D.; Tang, J.; Cao, W.; Lu, Z.; Cheng, Q.; Yang, J.; Bai, J.; Zhang, Y.; et al. Rap2B promotes proliferation, migration, and invasion of human breast cancer through calcium-related ERK1/2 signaling pathway. Sci. Rep. 2015, 5, 12363. [Google Scholar] [CrossRef] [PubMed]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Rybarczyk, P.; Sahni, J.; Sevestre, H.; Scharenberg, A.M.; Ouadid-Ahidouch, H. Transient receptor potential melastatin 7 is involved in oestrogen receptor-negative metastatic breast cancer cells migration through its kinase domain. Eur. J. Cancer 2013, 49, 3694–3707. [Google Scholar] [CrossRef] [PubMed]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.; Bravo-Cordero, J.J.; Condeelis, J.; Koleske, A.J.; Gil-Henn, H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011, 71, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.R.; Chen, C.L.; Chen, H.C. FAK is required for the assembly of podosome rosettes. J. Cell Biol. 2011, 195, 113–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Yoshida, S.; Muroi, E.; Yoshida, N.; Kawamura, M.; Kouchi, Z.; Nakamura, Y.; Sakai, R.; Fukami, K. Phosphoinositide 3-kinase signaling pathway mediated by p110alpha regulates invadopodia formation. J. Cell Biol. 2011, 193, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelassi, B.; Chantome, A.; Alcaraz-Perez, F.; Baroja-Mazo, A.; Cayuela, M.L.; Pelegrin, P.; Surprenant, A.; Roger, S. P2X(7) receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 2011, 30, 2108–2122. [Google Scholar] [CrossRef] [Green Version]
- Parkash, J.A.I.; Asotra, K. Combinatorial intervention of prostaglandin E2 receptor and calcium sensing receptor to attenuate breast cancer cell proliferation, migration and bone metastasis. Exp. Ther. Med. 2010, 1, 227–231. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, M.C.; Peters, A.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Lu, S.; He, Z.; Huang, H.; Yao, Z.; Miao, Y.; Cai, C.; Zou, F. MCU-dependent negative sorting of miR-4488 to extracellular vesicles enhances angiogenesis and promotes breast cancer metastatic colonization. Oncogene 2020. [Google Scholar] [CrossRef]
- Sartorius, C.A.; Hanna, C.T.; Gril, B.; Cruz, H.; Serkova, N.J.; Huber, K.M.; Kabos, P.; Schedin, T.B.; Borges, V.F.; Steeg, P.S.; et al. Estrogen promotes the brain metastatic colonization of triple negative breast cancer cells via an astrocyte-mediated paracrine mechanism. Oncogene 2016, 35, 2881–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutter, F.; Holen, I.; Brown, H.K.; Cross, S.S.; Evans, C.A.; Walker, M.; Coleman, R.E.; Westbrook, J.A.; Selby, P.J.; Brown, J.E.; et al. Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: Evidence using a novel bone-seeking cell line. Endocr. Relat. Cancer 2014, 21, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Kiryushko, D.; Novitskaya, V.; Soroka, V.; Klingelhofer, J.; Lukanidin, E.; Berezin, V.; Bock, E. Molecular mechanisms of Ca2+ signaling in neurons induced by the S100A4 protein. Mol. Cell. Biol. 2006, 26, 3625–3638. [Google Scholar] [CrossRef] [Green Version]
- Sherbet, G.V. Metastasis promoter S100A4 is a potentially valuable molecular target for cancer therapy. Cancer Lett. 2009, 280, 15–30. [Google Scholar] [CrossRef]
- Resende, R.R.; Andrade, L.M.; Oliveira, A.G.; Guimaraes, E.S.; Guatimosim, S.; Leite, M.F. Nucleoplasmic calcium signaling and cell proliferation: Calcium signaling in the nucleus. Cell Commun. Signal. 2013, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, E.; Machado, R.; Fonseca, M.C.; Franca, A.; Carvalho, C.; Araujo, E.S.A.C.; Almeida, B.; Cassini, P.; Hissa, B.; Drumond, L.; et al. Inositol 1, 4, 5-trisphosphate-dependent nuclear calcium signals regulate angiogenesis and cell motility in triple negative breast cancer. PLoS ONE 2017, 12, e0175041. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Liang, Y.; Diao, X.; Chen, Q. S100A4 promotes invasion and angiogenesis in breast cancer MDA-MB-231 cells by upregulating matrix metalloproteinase-13. Acta Biochim. Polocy 2012, 59, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.G.; Meng, Q.; Qi, F.M.; Yang, Q.F. Blocking TGF-beta inhibits breast cancer cell invasiveness via ERK/S100A4 signal. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3844–3853. [Google Scholar] [PubMed]
- Qu, S.; Wu, J.; Bao, Q.; Yao, B.; Duan, R.; Chen, X.; Li, L.; Yuan, H.; Jin, Y.; Ma, C. Osterix promotes the migration and angiogenesis of breast cancer by upregulation of S100A4 expression. J. Cell. Mol. Med. 2019, 23, 1116–1127. [Google Scholar] [CrossRef]
- Yu, C.; Tang, W.; Wang, Y.; Shen, Q.; Wang, B.; Cai, C.; Meng, X.; Zou, F. Downregulation of ACE2/Ang-(1-7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett. 2016, 376, 268–277. [Google Scholar] [CrossRef]
- Witzel, I.; Oliveira-Ferrer, L.; Pantel, K.; Muller, V.; Wikman, H. Breast cancer brain metastases: Biology and new clinical perspectives. Breast Cancer Res. 2016, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176. [Google Scholar] [CrossRef] [Green Version]
- Custodio-Santos, T.; Videira, M.; Brito, M.A. Brain metastasization of breast cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 132–147. [Google Scholar] [CrossRef]
- Lee, T.H.; Avraham, H.K.; Jiang, S.X.; Avraham, S. Vascular endothelial growth factor modulates the transendothelial migration of MDA-MB-231 breast cancer cells through regulation of brain microvascular endothelial cell permeability. J. Biol. Chem. 2003, 278, 5277–5284. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.C.; Lee, T.H.; Avraham, S.; Avraham, H.K. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res. 2004, 2, 327–338. [Google Scholar]
- Sharma, S.; Wu, S.Y.; Jimenez, H.; Xing, F.; Zhu, D.; Liu, Y.; Wu, K.; Tyagi, A.; Zhao, D.; Lo, H.W.; et al. Ca2+ and CACNA1H mediate targeted suppression of breast cancer brain metastasis by AM RF EMF. EBioMedicine 2019, 44, 194–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, B.J.; Buttery, R.; Lad, Y.; Banks, S.; Haslett, C.; Sethi, T. Integrin activation by Fam38A uses a novel mechanism of R-Ras targeting to the endoplasmic reticulum. J. Cell Sci. 2010, 123, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Pathak, M.M.; Nourse, J.L.; Tran, T.; Hwe, J.; Arulmoli, J.; Le, D.T.; Bernardis, E.; Flanagan, L.A.; Tombola, F. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 16148–16153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.B.; Morcos, P.A.; Rosenblatt, J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef]
- Hung, W.C.; Yang, J.R.; Yankaskas, C.L.; Wong, B.S.; Wu, P.H.; Pardo-Pastor, C.; Serra, S.A.; Chiang, M.J.; Gu, Z.; Wirtz, D.; et al. Confinement Sensing and Signal Optimization via Piezo1/PKA and Myosin II Pathways. Cell Rep. 2016, 15, 1430–1441. [Google Scholar] [CrossRef] [Green Version]
- Pardo-Pastor, C.; Rubio-Moscardo, F.; Vogel-Gonzalez, M.; Serra, S.A.; Afthinos, A.; Mrkonjic, S.; Destaing, O.; Abenza, J.F.; Fernandez-Fernandez, J.M.; Trepat, X.; et al. Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses. Proc. Natl. Acad. Sci. USA 2018, 115, 1925–1930. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Zhu, X.; Kong, R.; Zhao, Z.; Sheng, J.; Wang, J.; Xu, X.; Liu, J.; Cui, K.; Zhang, X.H.; et al. Targeting Brain-Adaptive Cancer Stem Cells Prohibits Brain Metastatic Colonization of Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 2052–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ovejero, D.; Veiga, S.; Garcia-Segura, L.M.; Doncarlos, L.L. Glial expression of estrogen and androgen receptors after rat brain injury. J. Comp. Neurol. 2002, 450, 256–271. [Google Scholar] [CrossRef] [Green Version]
- Fares, H.; DiNicolantonio, J.J.; O’Keefe, J.H.; Lavie, C.J. Amlodipine in hypertension: A first-line agent with efficacy for improving blood pressure and patient outcomes. Open Heart 2016, 3, e000473. [Google Scholar] [CrossRef] [Green Version]
- Hansson, L.; Hedner, T.; Lund-Johansen, P.; Kjeldsen, S.E.; Lindholm, L.H.; Syvertsen, J.O.; Lanke, J.; de Faire, U.; Dahlof, B.; Karlberg, B.E. Randomised trial of effects of calcium antagonists compared with diuretics and beta-blockers on cardiovascular morbidity and mortality in hypertension: The Nordic Diltiazem (NORDIL) study. Lancet 2000, 356, 359–365. [Google Scholar] [CrossRef]
- Hornung, R.S.; Jones, R.I.; Gould, B.A.; Sonecha, T.; Raftery, E.B. Twice-daily verapamil for hypertension: A comparison with propranolol. Am. J. Cardiol. 1986, 57, 93D–98D. [Google Scholar] [CrossRef]
- Taylor, J.M.; Simpson, R.U. Inhibition of cancer cell growth by calcium channel antagonists in the athymic mouse. Cancer Res. 1992, 52, 2413–2418. [Google Scholar]
- Brogden, R.N.; Markham, A. Mibefradil. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in the management of hypertension and angina pectoris. Drugs 1997, 54, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; McMonagle, T. Pimozide for schizophrenia or related psychoses. Cochrane Database Syst. Rev. 2000, 10. [Google Scholar] [CrossRef]
- Bertolesi, G.E.; Shi, C.; Elbaum, L.; Jollimore, C.; Rozenberg, G.; Barnes, S.; Kelly, M.E. The Ca2+ channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol. Pharmacol. 2002, 62, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, R.; Fruasaha, P.; Kozak, J.A. 2-Aminoethyl diphenyl borinate (2-APB) inhibits TRPM7 channels through an intracellular acidification mechanism. Channels 2012, 6, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dilger, J.P.; Lin, J. The Role of Transient Receptor Potential Melastatin 7 (TRPM7) in Cell Viability: A Potential Target to Suppress Breast Cancer Cell Cycle. Cancers 2020, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Fliegert, R.; Guse, A.H.; Lu, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85. [Google Scholar] [CrossRef]
- Bae, S.H.; Park, J.H.; Choi, H.G.; Kim, H.; Kim, S.H. Imidazole Antifungal Drugs Inhibit the Cell Proliferation and Invasion of Human Breast Cancer Cells. Biomol. Ther. 2018, 26, 494–502. [Google Scholar] [CrossRef]
- Guo, L.; Li, Z.S.; Wang, H.L.; Ye, C.Y.; Zhang, D.C. Carboxyamido-triazole inhibits proliferation of human breast cancer cells via G2/M cell cycle arrest and apoptosis. Eur. J. Pharmacol. 2006, 538, 15–22. [Google Scholar] [CrossRef]
- Zhang, B.B.; Wang, D.G.; Guo, F.F.; Xuan, C. Mitochondrial membrane potential and reactive oxygen species in cancer stem cells. Fam. Cancer 2015, 14, 19–23. [Google Scholar] [CrossRef]
- Kotnova, A.P.; Lyanova, B.M.; Dukhanina, E.A.; Portseva, T.N.; Ilyin, Y.V.; Georgieva, S.G.; Stepchenko, A.G.; Pankratova, E.V. Thapsigargin, Inhibitor of Sarco-Endoplasmic Ca2+-ATPase, Effectively Suppresses the Expression of S100A4 Protein in Human Breast Cancer Cell Line. Dokl. Biochem. Biophys. 2019, 486, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, B.N.; Roh, J.H.; An, Y.S.; Hur, H.; Yoon, J.K. Enhancing the Therapeutic Efficacy of 2-Deoxyglucose in Breast Cancer Cells Using Cell-cycle Synchronization. Anticancer Res. 2016, 36, 5975–5980. [Google Scholar] [CrossRef]
- Motawi, T.M.; Sadik, N.A.; Fahim, S.A.; Shouman, S.A. Combination of imatinib and clotrimazole enhances cell growth inhibition in T47D breast cancer cells. Chem. Biol. Interact. 2015, 233, 147–156. [Google Scholar] [CrossRef]
- Kim, P.K.; Zamora, R.; Petrosko, P.; Billiar, T.R. The regulatory role of nitric oxide in apoptosis. Int. Immunopharmacol. 2001, 1, 1421–1441. [Google Scholar] [CrossRef]
- Maes, M.; Vanhaecke, T.; Cogliati, B.; Yanguas, S.C.; Willebrords, J.; Rogiers, V.; Vinken, M. Measurement of Apoptotic and Necrotic Cell Death in Primary Hepatocyte Cultures. Methods Mol. Biol. 2015, 1250, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lothstein, L.; Israel, M.; Sweatman, T.W. Anthracycline drug targeting: Cytoplasmic versus nuclear--a fork in the road. Drug Resist Updat 2001, 4, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase poisons: Harnessing the dark side of enzyme mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [Google Scholar] [CrossRef] [Green Version]
- Frishman, W.H.; Sung, H.M.; Yee, H.C.M.; Liu, L.L.; Einzig, A.I.; Dutcher, J.; Keefe, D. Cardiovascular toxicity with cancer chemotherapy—Introduction. Curr. Prob. Cardiol. 1996, 21, 227–286. [Google Scholar] [CrossRef]
- Olson, R.D.; Mushlin, P.S. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J. 1990, 4, 3076–3086. [Google Scholar] [CrossRef]
- Minotti, G.; Licata, S.; Saponiero, A.; Menna, P.; Calafiore, A.M.; Di Giammarco, G.; Liberi, G.; Animati, F.; Cipollone, A.; Manzini, S.; et al. Anthracycline metabolism and toxicity in human myocardium: Comparisons between doxorubicin, epirubicin, and a novel disaccharide analogue with a reduced level of formation and [4Fe-4S] reactivity of its secondary alcohol metabolite. Chem. Res. Toxicol. 2000, 13, 1336–1341. [Google Scholar] [CrossRef]
- Ravid, A.; Rocker, D.; Machlenkin, A.; Rotem, C.; Hochman, A.; Kessler-Icekson, G.; Liberman, U.A.; Koren, R. 1,25-Dihydroxyvitamin D3 enhances the susceptibility of breast cancer cells to doxorubicin-induced oxidative damage. Cancer Res. 1999, 59, 862–867. [Google Scholar]
- Al-Malky, H.S.; Osman, A.M.; Damanhouri, Z.A.; Alkreathy, H.M.; Al Aama, J.Y.; Ramadan, W.S.; Al Qahtani, A.A.; Al Mahdi, H.B. Modulation of doxorubicin-induced expression of the multidrug resistance gene in breast cancer cells by diltiazem and protection against cardiotoxicity in experimental animals. Cancer Cell Int. 2019, 19, 191. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.J.; Ying, X.; Tian, W.; Yao, H.J.; Men, Y.; Yu, Y.; Zhang, L.A.; Ju, R.J.; Wang, X.X.; et al. Targeting Therapy with Mitosomal Daunorubicin plus Amlodipine Has the Potential To Circumvent Intrinsic Resistant Breast Cancer. Mol. Pharmaceut. 2011, 8, 162–175. [Google Scholar] [CrossRef]
- Diver, J.M.; Sage, S.O.; Rosado, J.A. The inositol trisphosphate receptor antagonist 2-aminoethoxydiphenylborate (2-APB) blocks Ca2+ entry channels in human platelets: Cautions for its use in studying Ca2+ influx. Cell Calcium 2001, 30, 323–329. [Google Scholar] [CrossRef]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Zeng, F.; Boulay, G.; Grimm, C.; Harteneck, C.; Beech, D.J. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 2005, 145, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19, 2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.D.; Hanley, P.J.; Rinne, S.; Zuzarte, M.; Daut, J. Calcium-activated K+ channel (K(Ca)3.1) activity during Ca2+ store depletion and store-operated Ca2+ entry in human macrophages. Cell Calcium 2010, 48, 19–27. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.; Hong, J.H. Ca2+ Signaling as the Untact Mode during Signaling in Metastatic Breast Cancer. Cancers 2021, 13, 1473. https://doi.org/10.3390/cancers13061473
Lee D, Hong JH. Ca2+ Signaling as the Untact Mode during Signaling in Metastatic Breast Cancer. Cancers. 2021; 13(6):1473. https://doi.org/10.3390/cancers13061473
Chicago/Turabian StyleLee, Dongun, and Jeong Hee Hong. 2021. "Ca2+ Signaling as the Untact Mode during Signaling in Metastatic Breast Cancer" Cancers 13, no. 6: 1473. https://doi.org/10.3390/cancers13061473