
The Potential Equivalents of TET2 Mutations




{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
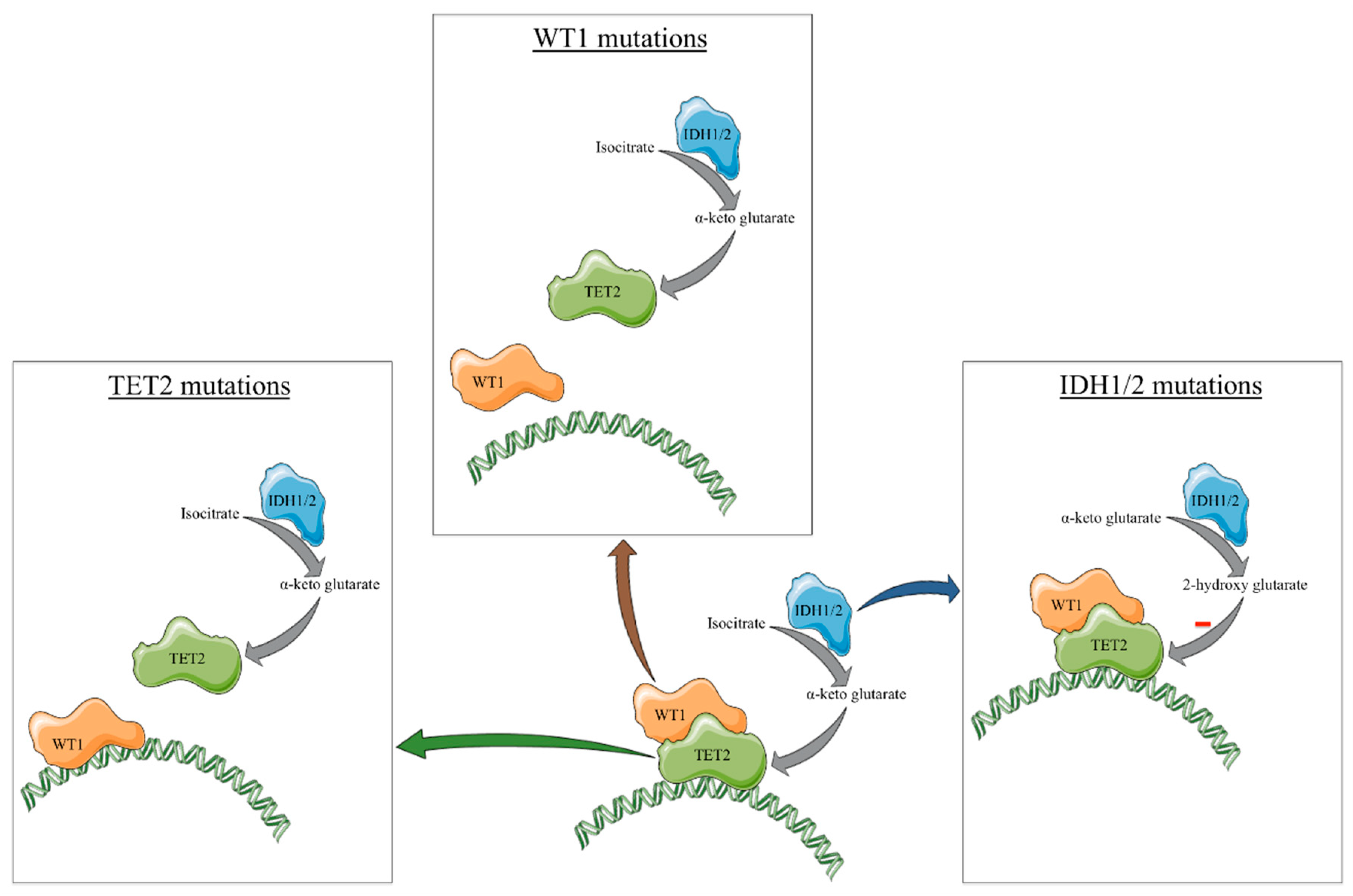
2. Classic Gene Mutations Mimicking TET2 Loss
3. Interactions within the TET2/IDH1/2/WT1 Mutated Cells
4. Other Equivalents of TET2 Mutations
4.1. Transcription Alteration
4.2. miRs
4.3. Direct Interaction
4.4. Posttranslational Changes
4.5. Substrate Reduction
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 2019, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, V.; Fernández, A.F.; Fraga, M.F. The Role of 5-Hydroxymethylcytosine in Development, Aging and Age-Related Diseases. Ageing Res. Rev. 2017, 37, 28–38. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, K.; Wang, L.; Zhu, Y.; Zhou, G.; Xie, D.; Yang, Q. IDH1/2 Mutants Inhibit TET-Promoted Oxidation of RNA 5mC to 5hmC. PLoS ONE 2016, 11, e0161261. [Google Scholar] [CrossRef] [Green Version]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) Gene in Hematopoiesis and Hematopoietic Diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Pronier, E.; Almire, C.; Mokrani, H.; Vasanthakumar, A.; Simon, A.; da Costa Reis Monte Mor, B.; Massé, A.; Le Couédic, J.-P.; Pendino, F.; Carbonne, B.; et al. Inhibition of TET2-Mediated Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine Disturbs Erythroid and Granulomonocytic Differentiation of Human Hematopoietic Progenitors. Blood 2011, 118, 2551–2555. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Zhang, R.-P.; Wan, F.; Guo, D.-Y.; Wang, P.; Xiang, L.-X.; Shao, J.-Z. TET2 Plays an Essential Role in Erythropoiesis by Regulating Lineage-Specific Genes via DNA Oxidative Demethylation in a Zebrafish Model. Mol. Cell. Biol. 2014, 34, 989–1002. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Cai, X.; Cai, C.-L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.-C.; Xu, M. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor Development Is Associated with Decrease of TET Gene Expression and 5-Methylcytosine Hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A Targeted Mutational Landscape of Angioimmunoblastic T-Cell Lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef]
- Yao, W.-Q.; Wu, F.; Zhang, W.; Chuang, S.-S.; Thompson, J.S.; Chen, Z.; Zhang, S.-W.; Clipson, A.; Wang, M.; Liu, H.; et al. Angioimmunoblastic T-Cell Lymphoma Contains Multiple Clonal T-Cell Populations Derived from a Common TET2 Mutant Progenitor Cell. J. Pathol. 2020, 250, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Scherm, M.G.; Serr, I.; Zahm, A.M.; Schug, J.; Bellusci, S.; Manfredini, R.; Salb, V.K.; Gerlach, K.; Weigmann, B.; Ziegler, A.-G.; et al. MiRNA142-3p Targets Tet2 and Impairs Treg Differentiation and Stability in Models of Type 1 Diabetes. Nat. Commun. 2019, 10, 5697. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Lio, C.-W.J.; Samaniego-Castruita, D.; Li, X.; Rao, A. Loss of TET2 and TET3 in Regulatory T Cells Unleashes Effector Function. Nat. Commun. 2019, 10, 2011. [Google Scholar] [CrossRef] [Green Version]
- Cortés, J.R.; Palomero, T. The Curious Origins of Angioimmunoblastic T-Cell Lymphoma. Curr. Opin. Hematol. 2016, 23, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venneker, S.; Szuhai, K.; Hogendoorn, P.C.W.; Bovée, J.V.M.G. Mutation-Driven Epigenetic Alterations as a Defining Hallmark of Central Cartilaginous Tumours, Giant Cell Tumour of Bone and Chondroblastoma. Virchows Arch. 2020, 476, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-H.; Pierscianek, D.; Mittelbronn, M.; Vital, A.; Mariani, L.; Hasselblatt, M.; Ohgaki, H. TET2 Promoter Methylation in Low-Grade Diffuse Gliomas Lacking IDH1/2 Mutations: Figure 1. J. Clin. Pathol. 2011, 64, 850–852. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Pasca, S.; Turcas, C.; Jurj, A.; Teodorescu, P.; Iluta, S.; Hotea, I.; Bojan, A.; Selicean, C.; Fetica, B.; Petrushev, B.; et al. The Influence of Methylating Mutations on Acute Myeloid Leukemia: Preliminary Analysis on 56 Patients. Diagnostics 2020, 10, 263. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Lu, C.; Cross, J.R.; Morris, J.P.; Shroff, A.S.; Ward, P.S.; Bradner, J.E.; Thompson, C.; Lowe, S.W. Cancer-Associated IDH2 Mutants Drive an Acute Myeloid Leukemia That Is Susceptible to Brd4 Inhibition. Genes Dev. 2013, 27, 1974–1985. [Google Scholar] [CrossRef] [Green Version]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1R132H in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Venneti, S.; Akalin, A.; Fang, F.; Ward, P.S.; DeMatteo, R.G.; Intlekofer, A.M.; Chen, C.; Ye, J.; Hameed, M.; et al. Induction of Sarcomas by Mutant IDH2. Genes Dev. 2013, 27, 1986–1998. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH Inhibits HNF-4α to Block Hepatocyte Differentiation and Promote Biliary Cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.-É.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib Induces Deep Durable Remissions in Patients with Newly Diagnosed IDH1-Mutant Acute Myeloid Leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, O.A.; Falchi, L.; Lue, J.K.; Marchi, E.; Kinahan, C.; Sawas, A.; Deng, C.; Montanari, F.; Amengual, J.E.; Kim, H.A.; et al. Oral 5-Azacytidine and Romidepsin Exhibit Marked Activity in Patients with PTCL: A Multicenter Phase 1 Study. Blood 2019, 134, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, F.; Dupuis, J.; Sujobert, P.; Tournillhac, O.; Cheminant, M.; Sarkozy, C.; Pelletier, L.; Marçais, A.; Robe, C.; Fataccioli, V.; et al. Treatment with 5-Azacytidine Induces a Sustained Response in Patients with Angioimmunoblastic T-Cell Lymphoma. Blood 2018, 132, 2305–2309. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in Hematopoietic Cells Leads to DNA Hypermethylation of Active Enhancers and Induction of Leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) Mutation Increases Murine Haematopoietic Progenitors and Alters Epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joberty, G.; Boesche, M.; Brown, J.A.; Eberhard, D.; Garton, N.S.; Humphreys, P.G.; Mathieson, T.; Muelbaier, M.; Ramsden, N.G.; Reader, V.; et al. Interrogating the Druggability of the 2-Oxoglutarate-Dependent Dioxygenase Target Class by Chemical Proteomics. ACS Chem. Biol. 2016, 11, 2002–2010. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Makishima, H.; Tiu, R.V.; Szpurka, H.; Huang, Y.; Traina, F.; Visconte, V.; Sugimoto, Y.; Prince, C.; O’Keefe, C.; et al. Mutational Spectrum Analysis of Chronic Myelomonocytic Leukemia Includes Genes Associated with Epigenetic Regulation: UTX, EZH2, and DNMT3A. Blood 2011, 118, 3932–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Lemonnier, F.; Mak, T.W. Roles of IDH1/2 and TET2 Mutations in Myeloid Disorders. Int. J. Hematol. 2016, 103, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent Somatic TET2 Mutations in Normal Elderly Individuals with Clonal Hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Zheng, G.; Chen, P.; Pallavajjalla, A.; Haley, L.; Gondek, L.; Dezern, A.; Ling, H.; De Marchi, F.; Lin, M.; Gocke, C. The Diagnostic Utility of Targeted Gene Panel Sequencing in Discriminating Etiologies of Cytopenia. Am. J. Hematol. 2019, 94, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C. What Do We Know about IDH1/2 Mutations so Far, and How Do We Use It? Acta Neuropathol. 2013, 125, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Abbas, S.; Erpelinck-Verschueren, C.A.J.; Goudswaard, C.S.; Löwenberg, B.; Valk, P.J.M. Mutant Wilms’ Tumor 1 (WT1) MRNA with Premature Termination Codons in Acute Myeloid Leukemia (AML) Is Sensitive to Nonsense-Mediated RNA Decay (NMD). Leukemia 2010, 24, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xiao, M.; Chen, X.; Chen, L.; Xu, Y.; Lv, L.; Wang, P.; Yang, H.; Ma, S.; Lin, H.; et al. WT1 Recruits TET2 to Regulate Its Target Gene Expression and Suppress Leukemia Cell Proliferation. Mol. Cell 2015, 57, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauth, M.-T.; Alpermann, T.; Bacher, U.; Eder, C.; Dicker, F.; Ulke, M.; Kuznia, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; et al. WT1 Mutations Are Secondary Events in AML, Show Varying Frequencies and Impact on Prognosis between Genetic Subgroups. Leukemia 2015, 29, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.; Jurj, A.; Tomuleasa, C.; Zdrenghea, M. TET2/IDH1/2/WT1 and NPM1 Mutations Influence the RUNX1 Expression Correlations in Acute Myeloid Leukemia. Medicina 2020, 56, 637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.-H.; Rao, A.; et al. DNMT3A and TET2 Compete and Cooperate to Repress Lineage-Specific Transcription Factors in Hematopoietic Stem Cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Guo, Y.; Niu, Y.; Jin, J.; Zhang, X.; Shi, X.; Zhang, L.; Li, R.; Chen, L.; Ma, R.Z. Epigenetic Silencing of TET2 and TET3 Induces an EMT-like Process in Melanoma. Oncotarget 2017, 8, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhang, Q.; Wu, Q.; Wei, Y.; Yu, J.; Mu, J.; Zhang, J.; Zeng, W.; Feng, B. Effect of TET2 on the Pathogenesis of Diabetic Nephropathy through Activation of Transforming Growth Factor Β1 Expression via DNA Demethylation. Life Sci. 2018, 207, 127–137. [Google Scholar] [CrossRef]
- Chen, B.; Lei, Y.; Wang, H.; Dang, Y.; Fang, P.; Wang, J.; Yang, J.; Liu, L. Repression of the Expression of TET2 by ZEB1 Contributes to Invasion and Growth in Glioma Cells. Mol. Med. Rep. 2017, 15, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.P.; Miles, S.L. Silencing HIF-1α Induces TET2 Expression and Augments Ascorbic Acid Induced 5-Hydroxymethylation of DNA in Human Metastatic Melanoma Cells. Biochem. Biophys. Res. Commun. 2017, 490, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Luo, C.-W.; Lai, Y.-S.; Wu, C.-C.; Hung, W.-C. Lysine Demethylase KDM2A Inhibits TET2 to Promote DNA Methylation and Silencing of Tumor Suppressor Genes in Breast Cancer. Oncogenesis 2017, 6, e369. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zang, G.; Zhong, W.; Chen, R.; Zhang, Y.; Yang, P.; Yan, J. Activation of CD137 Signaling Promotes Neointimal Formation by Attenuating TET2 and Transferrring from Endothelial Cell-Derived Exosomes to Vascular Smooth Muscle Cells. Biomed. Pharmacother. 2020, 121, 109593. [Google Scholar] [CrossRef]
- Huang, F.; Sun, J.; Chen, W.; He, X.; Zhu, Y.; Dong, H.; Wang, H.; Li, Z.; Zhang, L.; Khaled, S.; et al. HDAC4 Inhibition Disrupts TET2 Function in High-Risk MDS and AML. Aging (Albany NY) 2020, 12, 16759–16774. [Google Scholar] [CrossRef]
- Noreen, F.; Küng, T.; Tornillo, L.; Parker, H.; Silva, M.; Weis, S.; Marra, G.; Rad, R.; Truninger, K.; Schär, P. DNA Methylation Instability by BRAF-Mediated TET Silencing and Lifestyle-Exposure Divides Colon Cancer Pathways. Clin. Epigenet. 2019, 11, 196. [Google Scholar] [CrossRef]
- Hamberg, M.; Backes, C.; Fehlmann, T.; Hart, M.; Meder, B.; Meese, E.; Keller, A. MiRTargetLink—MiRNAs, Genes and Interaction Networks. Int. J. Mol. Sci. 2016, 17, 564. [Google Scholar] [CrossRef]
- Fan, Y.; Xia, J. miRNet—Functional Analysis and Visual Exploration of miRNA–Target Interactions in a Network Context. In Computational Cell Biology; Methods in Molecular Biology; von Stechow, L., Santos Delgado, A., Eds.; Springer: New York, NY, USA, 2018; Volume 1819, pp. 215–233. ISBN 978-1-4939-8617-0. [Google Scholar]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA–Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhu, Y.; He, X.; Ding, Z.; Dong, H.; Feng, Y.; Yu, X.; Zhao, D.; Wu, H.; Feng, L.; et al. Activation of SIRT1 Deacetylase As a Therapeutic Approach for Myelodysplastic Syndromes By Restoring TET2 Function. Blood 2017, 130. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The Oncogenic MicroRNA MiR-22 Targets the TET2 Tumor Suppressor to Promote Hematopoietic Stem Cell Self-Renewal and Transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Xia, L.; Fan, S.; Zheng, J.; Qin, J.; Fan, X.; Liu, Y.; Tao, J.; Liu, Y.; Li, K.; et al. Circular RNA CircMAP3K5 Acts as a MicroRNA-22-3p Sponge to Promote Resolution of Intimal Hyperplasia via TET2-Mediated SMC Differentiation. Circulation 2021, 143, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Fráguas, M.S.; Eggenschwiler, R.; Hoepfner, J.; dos Santos Schiavinato, J.L.; Haddad, R.; Oliveira, L.H.B.; Araújo, A.G.; Zago, M.A.; Panepucci, R.A.; Cantz, T. MicroRNA-29 Impairs the Early Phase of Reprogramming Process by Targeting Active DNA Demethylation Enzymes and Wnt Signaling. Stem Cell Res. 2017, 19, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cao, Y.; Zhai, Y.; Ma, X.; An, X.; Zhang, S.; Li, Z. MicroRNA-29b Regulates DNA Methylation by Targeting Dnmt3a/3b and Tet1/2/3 in Porcine Early Embryo Development. Dev. Growth Differ. 2018, 60, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Li, X.; Kong, L.; Xu, Q.; Wang, Z.; Lv, Q. MiR-101-3p Induces Vascular Endothelial Cell Dysfunction by Targeting Tet Methylcytosine Dioxygenase 2. Acta Biochim. Biophys. Sin. 2020, 52, 180–191. [Google Scholar] [CrossRef]
- Liu, J.; Guo, B.; Chen, Z.; Wang, N.; Iacovino, M.; Cheng, J.; Roden, C.; Pan, W.; Khan, S.; Chen, S.; et al. MiR-125b Promotes MLL-AF9–Driven Murine Acute Myeloid Leukemia Involving a VEGFA-Mediated Non–Cell-Intrinsic Mechanism. Blood 2017, 129, 1491–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Li, D.-W.; Wang, S.-N.; Luo, S.; Li, Q.; Huang, P.; Wang, J.-M.; Xu, M.; Xu, C.-X. MiR-125a Promotes the Progression of Giant Cell Tumors of Bone by Stimulating IL-17A and β-Catenin Expression. Mol. Ther. Nucleic Acids 2018, 13, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Zhaolin, Z.; Jiaojiao, C.; Peng, W.; Yami, L.; Tingting, Z.; Jun, T.; Shiyuan, W.; Jinyan, X.; Dangheng, W.; Zhisheng, J.; et al. OxLDL Induces Vascular Endothelial Cell Pyroptosis through MiR-125a-5p/TET2 Pathway. J. Cell. Physiol. 2019, 234, 7475–7491. [Google Scholar] [CrossRef]
- Ren, S.; Xu, Y. AC016405.3, a Novel Long Noncoding RNA, Acts as a Tumor Suppressor through Modulation of TET2 by MicroRNA-19a-5p Sponging in Glioblastoma. Cancer Sci. 2019, 110, 1621–1632. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, L.; Liu, Y.; Luo, S.; Song, Y.; Fang, B. MiR-144-3p Suppresses Osteogenic Differentiation of BMSCs from Patients with Aplastic Anemia through Repression of TET2. Mol. Ther. Nucleic Acids 2020, 19, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-B.; Jia, B.; Wang, W.; Xu, G.-H.; Guo, J.-C.; Li, X.; Liu, J.-N. Role of MiR-199a-5p in Osteoblast Differentiation by Targeting TET2. Gene 2020, 726, 144193. [Google Scholar] [CrossRef]
- Peng, B.; Li, C.; He, L.; Tian, M.; Li, X. MiR-660-5p Promotes Breast Cancer Progression through down-Regulating TET2 and Activating PI3K/AKT/MTOR Signaling. Braz. J. Med. Biol. Res. 2020, 53, e9740. [Google Scholar] [CrossRef]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Äijö, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lähdesmäki, H.; et al. Modulation of TET2 Expression and 5-Methylcytosine Oxidation by the CXXC Domain Protein IDAX. Nature 2013, 497, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Guilhamon, P.; Eskandarpour, M.; Halai, D.; Wilson, G.A.; Feber, A.; Teschendorff, A.E.; Gomez, V.; Hergovich, A.; Tirabosco, R.; Fernanda Amary, M.; et al. Meta-Analysis of IDH-Mutant Cancers Identifies EBF1 as an Interaction Partner for TET2. Nat. Commun. 2013, 4, 2166. [Google Scholar] [CrossRef] [Green Version]
- Boller, S.; Li, R.; Grosschedl, R. Defining B Cell Chromatin: Lessons from EBF1. Trends Genet. 2018, 34, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Jimenez, N.; Sklias, A.; Ecsedi, S.; Cahais, V.; Degli-Esposti, D.; Jay, A.; Ancey, P.B.; Woo, H.D.; Hernandez-Vargas, H.; Herceg, Z. Lowly Methylated Region Analysis Identifies EBF1 as a Potential Epigenetic Modifier in Breast Cancer. Epigenetics 2017, 12, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Costa, Y.; Ding, J.; Theunissen, T.W.; Faiola, F.; Hore, T.A.; Shliaha, P.V.; Fidalgo, M.; Saunders, A.; Lawrence, M.; Dietmann, S.; et al. NANOG-Dependent Function of TET1 and TET2 in Establishment of Pluripotency. Nature 2013, 495, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, J.; Jin, Y.; Zheng, M.; Zhang, H.; Yuan, M.; Lv, Z.; Odhiambo, W.; Yu, X.; Zhang, P.; Li, C.; et al. AID and TET2 Co-Operation Modulates FANCA Expression by Active Demethylation in Diffuse Large B Cell Lymphoma: AID and TET2 Cooperation Modulates FANCA. Clin. Exp. Immunol. 2019, 195, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The Predator Becomes the Prey: Regulating the Ubiquitin System by Ubiquitylation and Degradation. Nat. Rev. Mol. Cell Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lv, L.; Nakagawa, M.; Yu, Y.; Yu, C.; D’Alessio, A.C.; Nakayama, K.; Fan, H.-Y.; Chen, X.; Xiong, Y. CRL4VprBP E3 Ligase Promotes Monoubiquitylation and Chromatin Binding of TET Dioxygenases. Mol. Cell 2015, 57, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Smith, M.D.; Lv, L.; Nakagawa, T.; Li, Z.; Sun, S.-C.; Brown, N.G.; Xiong, Y.; Xu, Y. USP15 Suppresses Tumor Immunity via Deubiquitylation and Inactivation of TET2. Sci. Adv. 2020, 6, eabc9730. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 Restricts AKG Levels in AML Stem Cells Leading to IDHmut-like DNA Hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasca, S.; Jurj, A.; Zdrenghea, M.; Tomuleasa, C. The Potential Equivalents of TET2 Mutations. Cancers 2021, 13, 1499. https://doi.org/10.3390/cancers13071499
Pasca S, Jurj A, Zdrenghea M, Tomuleasa C. The Potential Equivalents of TET2 Mutations. Cancers. 2021; 13(7):1499. https://doi.org/10.3390/cancers13071499
Chicago/Turabian StylePasca, Sergiu, Ancuta Jurj, Mihnea Zdrenghea, and Ciprian Tomuleasa. 2021. "The Potential Equivalents of TET2 Mutations" Cancers 13, no. 7: 1499. https://doi.org/10.3390/cancers13071499
APA StylePasca, S., Jurj, A., Zdrenghea, M., & Tomuleasa, C. (2021). The Potential Equivalents of TET2 Mutations. Cancers, 13(7), 1499. https://doi.org/10.3390/cancers13071499