
Pulsed-Focused Ultrasound Slows B16 Melanoma and 4T1 Breast Tumor Growth through Differential Tumor Microenvironmental Changes

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Murine B16 and 4T1 Tumor Models
2.3. pFUS Treatment
2.4. Proteomic Analyses
2.5. Flow Cytometric Analyses
2.6. Immunohistochemistry Analysis (IHC)
2.7. RNA-Seq
2.8. Statistical Analyses
3. Results
3.1. pFUS Effect on Tumors
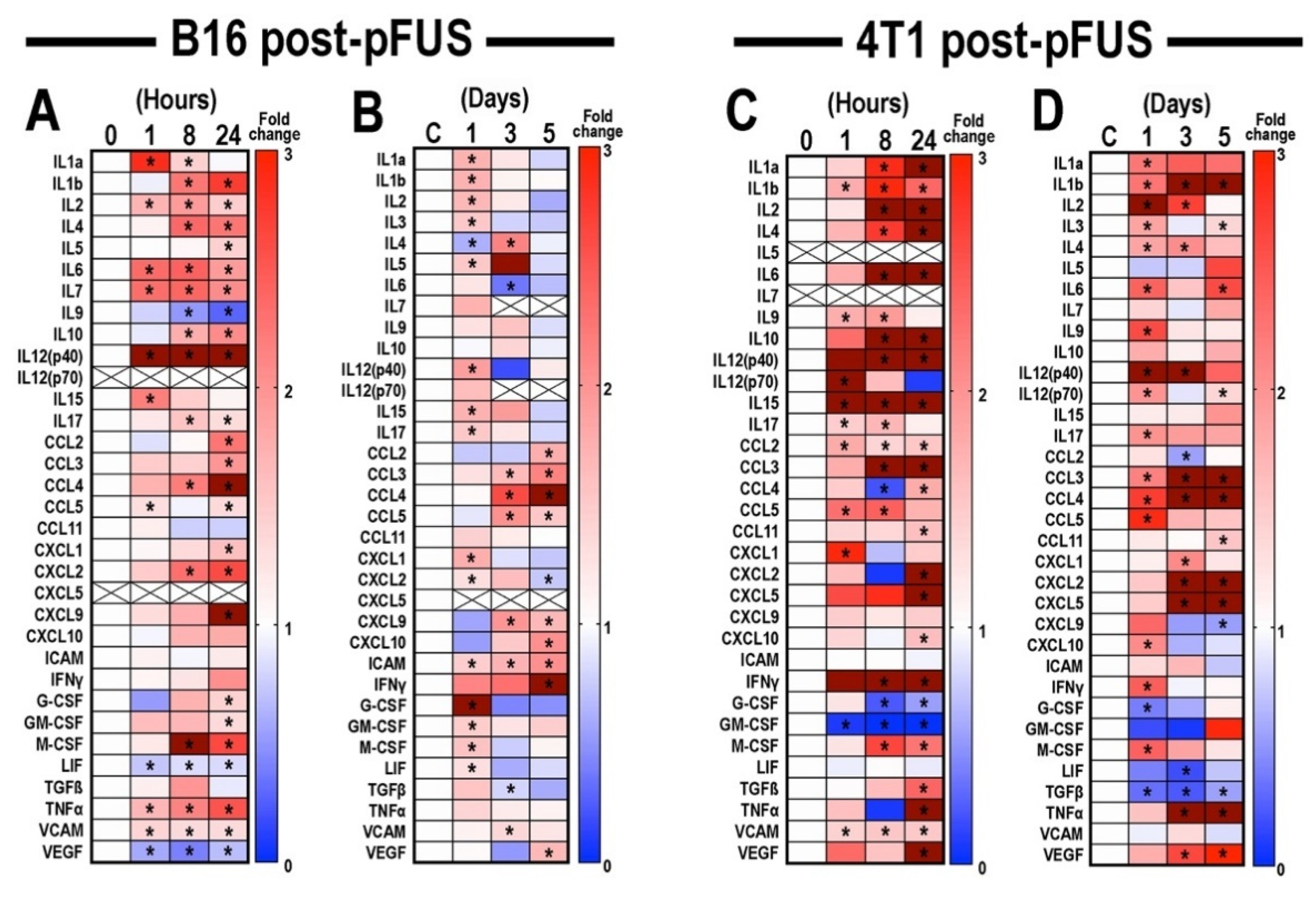
3.2. Proteomic Analyses in B16 and 4T1 Flank Tumors Following pFUS
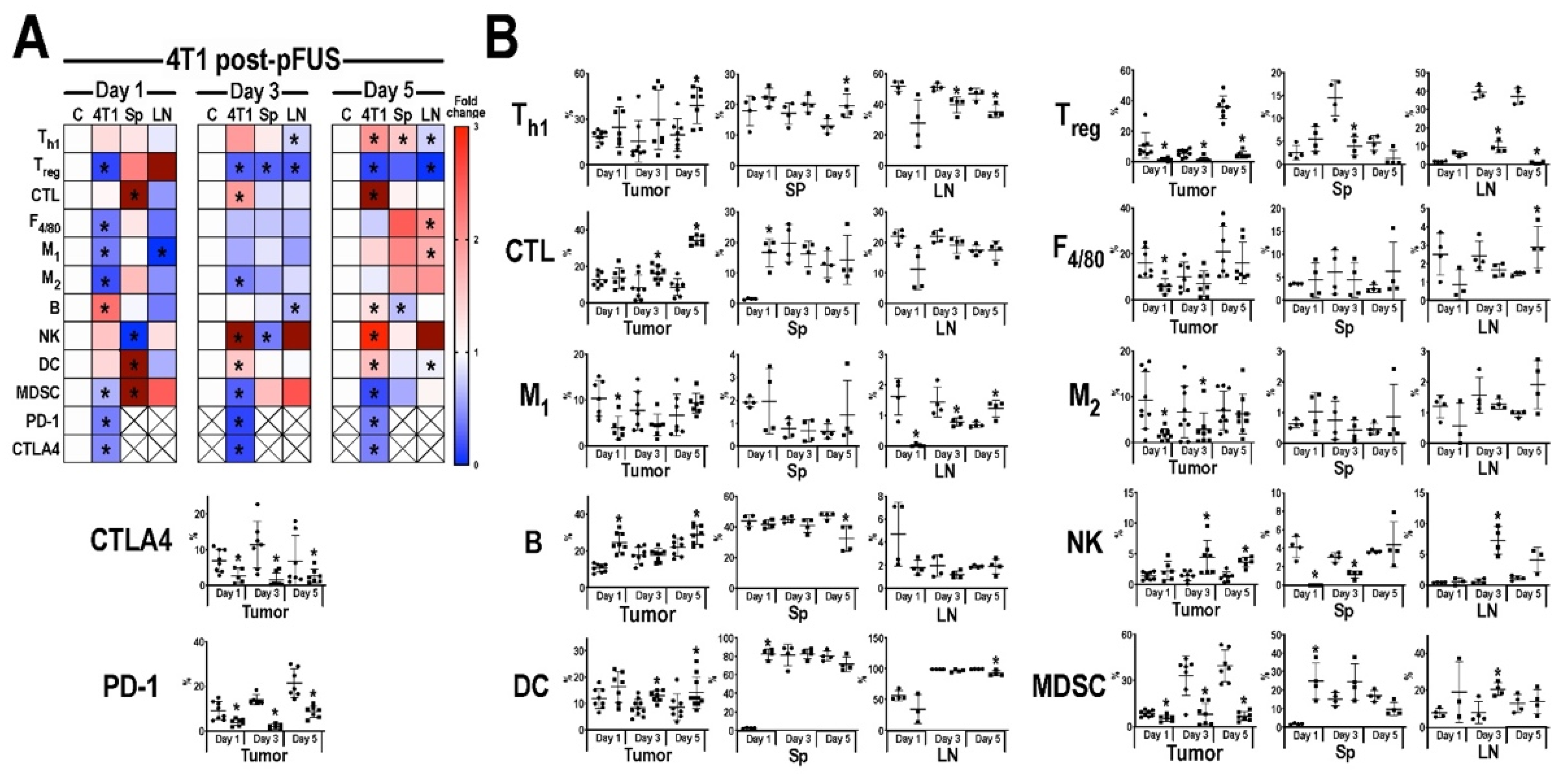
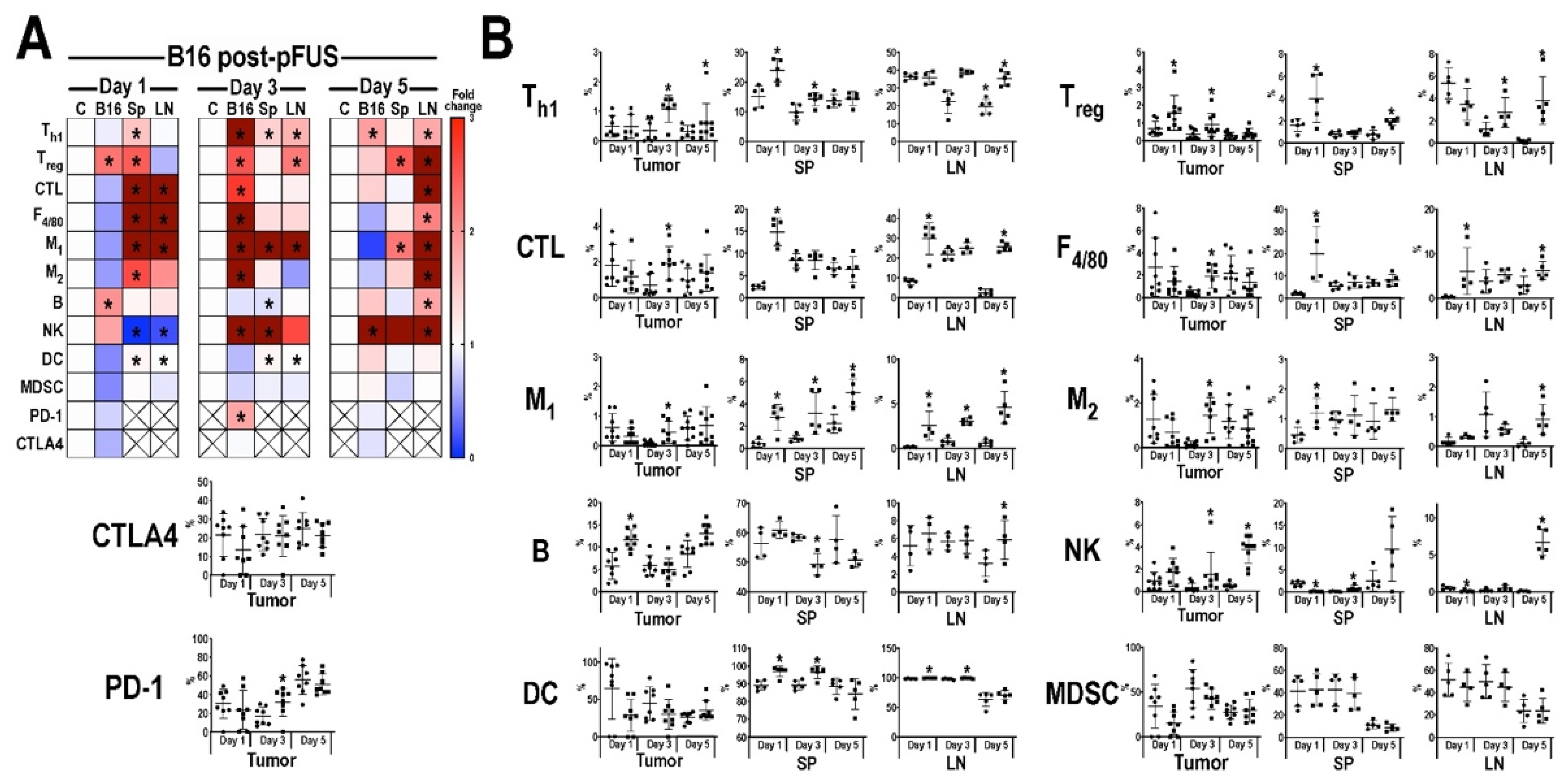
3.3. Flow Cytometric Analyses of Tumors, Inguinal Lymph Nodes, and Spleens Post-pFUS
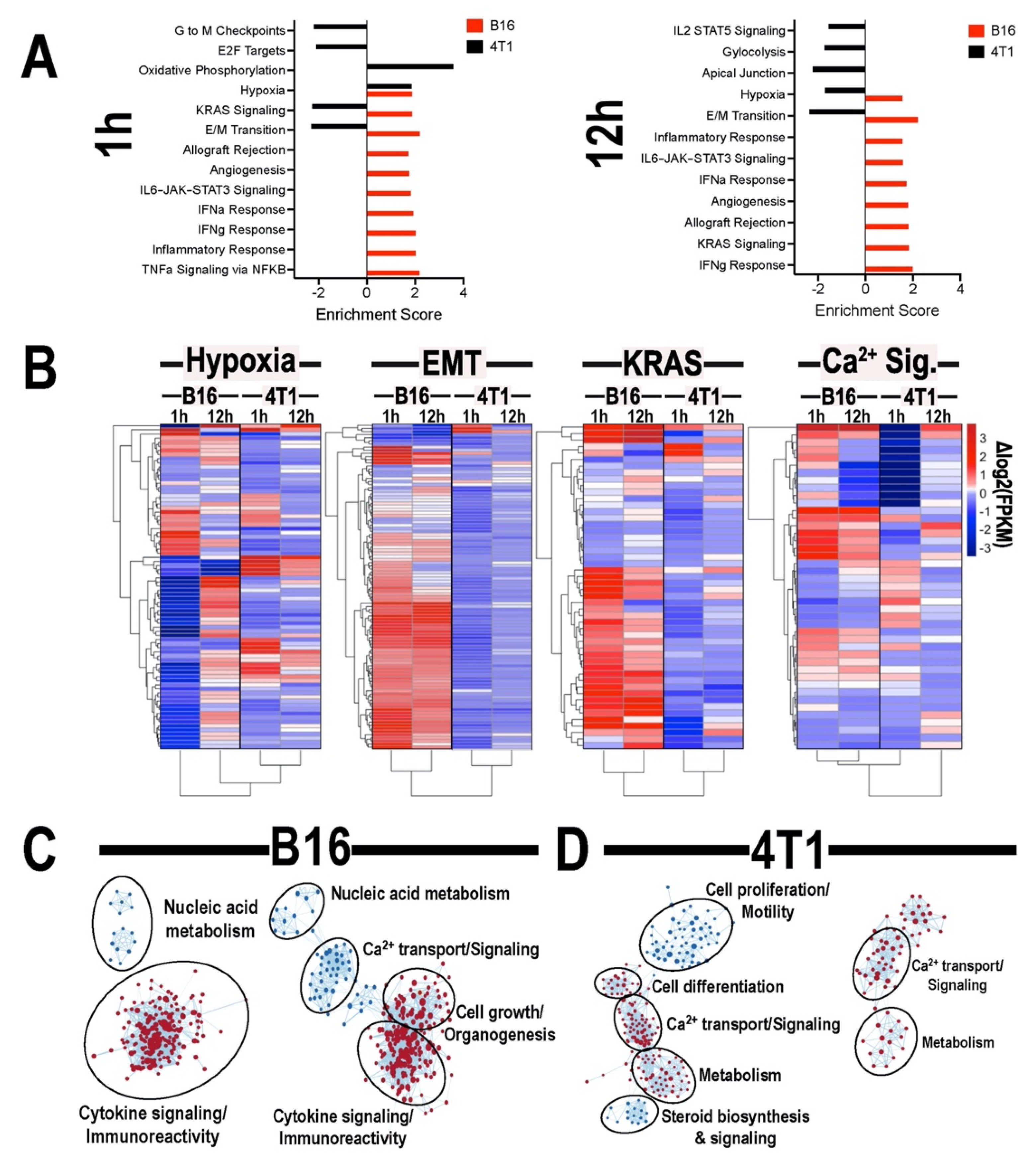
3.4. Transcriptomic Analyses of Tumors Following pFUS
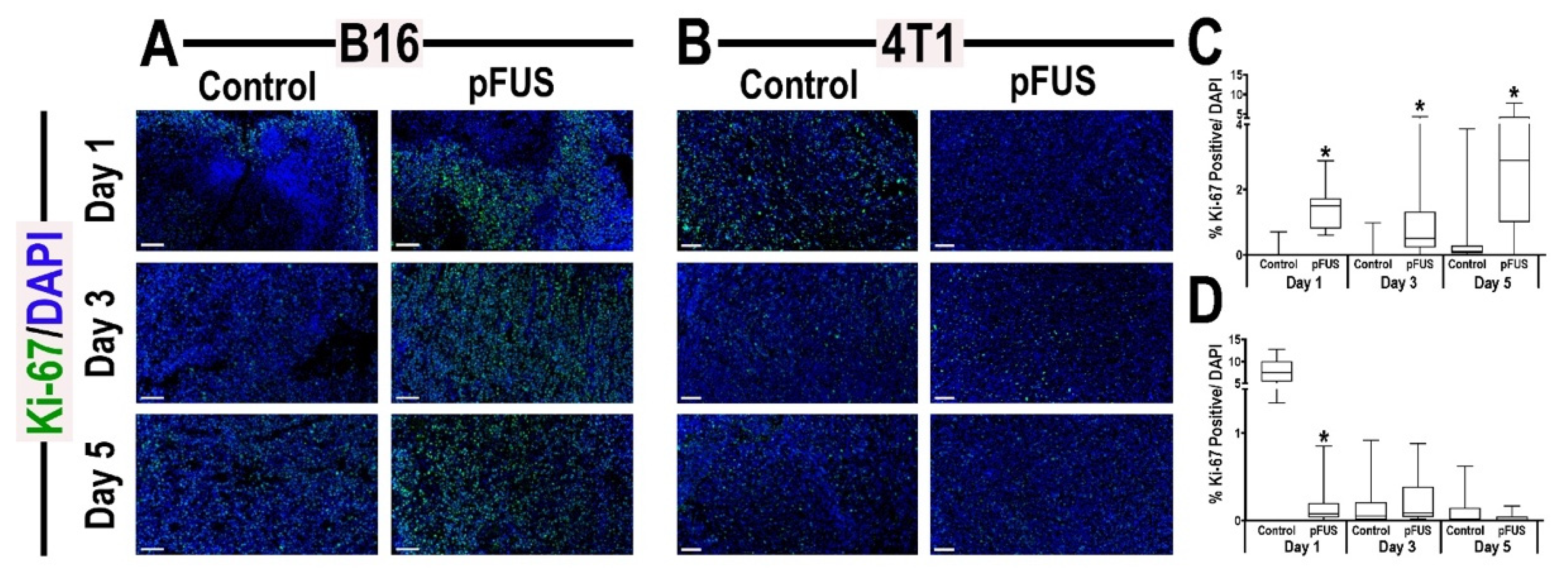
3.5. Histological Analyses of Tumors Following pFUS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol. 2019, 10, 1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Weiner, G.J. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Decade in review-cancer immunotherapy: Entering the mainstream of cancer treatment. Nat. Rev. Clin. Oncol. 2014, 11, 630–632. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Elhelf, I.A.S.; Albahar, H.; Shah, U.; Oto, A.; Cressman, E.; Almekkawy, M. High intensity focused ultrasound: The fundamentals, clinical applications and research trends. Diagn. Interv. Imaging 2018, 99, 349–359. [Google Scholar] [CrossRef]
- Silvestrini, M.T.; Ingham, E.S.; Mahakian, L.M.; Kheirolomoom, A.; Liu, Y.; Fite, B.Z.; Tam, S.M.; Tucci, S.T.; Watson, K.D.; Wong, A.W.; et al. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight 2017, 2, e90521. [Google Scholar] [CrossRef] [PubMed]
- Schade, G.R.; Wang, Y.N.; D’Andrea, S.; Hwang, J.H.; Liles, W.C.; Khokhlova, T.D. Boiling histotripsy ablation of renal cell carcinoma in the eker rat promotes a systemic inflammatory response. Ultrasound Med. Biol. 2019, 45, 137–147. [Google Scholar] [CrossRef]
- Qu, S.; Worlikar, T.; Felsted, A.E.; Ganguly, A.; Beems, M.V.; Hubbard, R.; Pepple, A.L.; Kevelin, A.A.; Garavaglia, H.; Dib, J.; et al. Non-thermal histotripsy tumor ablation promotes abscopal immune responses that enhance cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000200. [Google Scholar] [CrossRef] [Green Version]
- Mauri, G.; Nicosia, L.; Xu, Z.; Di Pietro, S.; Monfardini, L.; Bonomo, G.; Varano, G.M.; Prada, F.; Della Vigna, P.; Orsi, F. Focused ultrasound: Tumour ablation and its potential to enhance immunological therapy to cancer. Br. J. Radiol. 2018, 91, 20170641. [Google Scholar] [CrossRef]
- Burks, S.R.; Ziadloo, A.; Hancock, H.A.; Chaudhry, A.; Dean, D.D.; Lewis, B.K.; Frenkel, V.; Frank, J.A. Investigation of cellular and molecular responses to pulsed focused ultrasound in a mouse model. PLoS ONE 2011, 6, e24730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, K.W.; Tu, T.W.; Nagle, M.E.; Lewis, B.K.; Burks, S.R.; Frank, J.A. Molecular and histological effects of MR-guided pulsed focused ultrasound to the rat heart. J. Transl. Med. 2017, 15, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, Z.I.; Kim, S.; Jikaria, N.; Qureshi, F.; Milo, B.; Lewis, B.K.; Bresler, M.; Burks, S.R.; Frank, J.A. Disrupting the blood-brain barrier by focused ultrasound induces sterile inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E75–E84. [Google Scholar] [CrossRef] [Green Version]
- Burks, S.R.; Nguyen, B.A.; Tebebi, P.A.; Kim, S.J.; Bresler, M.N.; Ziadloo, A.; Street, J.M.; Yuen, P.S.; Star, R.A.; Frank, J.A. Pulsed focused ultrasound pretreatment improves mesenchymal stromal cell efficacy in preventing and rescuing established acute kidney injury in mice. Stem Cells 2015, 33, 1241–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, O.; Chandran, P.; Lorsung, R.R.; Cohen, G.; Burks, S.R.; Frank, J.A. The proteomic effects of pulsed focused ultrasound on tumor microenvironments of murine melanoma and breast cancer models. Ultrasound Med. Biol. 2019, 45, 3232–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G.; Chandran, P.; Lorsung, R.M.; Tomlinson, L.E.; Sundby, M.; Burks, S.R.; Frank, J.A. The impact of focused ultrasound in two tumor models: Temporal alterations in the natural history on tumor microenvironment and immune cell response. Cancers 2020, 12, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council (U.S.) Committee for the Update of the Guide for the Care and Use of Laboratory Animals; Institute for Laboratory Animal Research (U.S.). Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; 220p. [Google Scholar]
- Fleming, M.G.; Steger, C.; Zhang, J.; Gao, J.; Cognetta, A.B.; Pollak, I.; Dyer, C.R. Techniques for a structural analysis of dermatoscopic imagery. Comput. Med. Imaging Graph. 1998, 22, 375–389. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R.; Kulkarni, P. The genetic/non-genetic duality of drug resistance in cancer. Trends Cancer 2018, 4, 110–118. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Aleksakhina, S.N.; Kashyap, A.; Imyanitov, E.N. Mechanisms of acquired tumor drug resistance. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188310. [Google Scholar] [CrossRef]
- Eckstein, N.; Servan, K.; Hildebrandt, B.; Politz, A.; von Jonquieres, G.; Wolf-Kummeth, S.; Napierski, I.; Hamacher, A.; Kassack, M.U.; Budczies, J.; et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009, 69, 2996–3003. [Google Scholar] [CrossRef] [Green Version]
- Van den Bijgaart, R.J.; Eikelenboom, D.C.; Hoogenboom, M.; Futterer, J.J.; den Brok, M.H.; Adema, G.J. Thermal and mechanical high-intensity focused ultrasound: Perspectives on tumor ablation, immune effects and combination strategies. Cancer Immunol. Immunother. 2017, 66, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.L.; Hsieh, H.Y.; Lu, L.A.; Kang, C.W.; Wu, M.F.; Lin, C.Y. Low-pressure pulsed focused ultrasound with microbubbles promotes an anticancer immunological response. J. Transl. Med. 2012, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.L.; Zhu, X.Q.; Lu, P.; Zhou, Q.; Zhang, J.; Wu, F. Activation of tumor-infiltrating antigen presenting cells by high intensity focused ultrasound ablation of human breast cancer. Ultrasound Med. Biol. 2009, 35, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.W.; Seol, D.; Ding, L.; Lim, T.H.; Frank, J.A.; Martin, J.A. Ultrasound-mediated microbubble destruction suppresses melanoma tumor growth. Ultrasound Med. Biol. 2018, 44, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFbeta biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Cheung, K.; Ma, L.; Wang, G.; Coe, D.; Ferro, R.; Falasca, M.; Buckley, C.D.; Mauro, C.; Marelli-Berg, F.M. CD31 signals confer immune privilege to the vascular endothelium. Proc. Natl. Acad. Sci. USA 2015, 112, E5815–E5824. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felipe Lima, J.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J.M. EMT in breast carcinoma—A review. J. Clin. Med. 2016, 5, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaskovic-Crook, E.; Thompson, E.W.; Thiery, J.P. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 2009, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Durand, S.; Dalle, S.; Caramel, J. EMT-inducing transcription factors, drivers of melanoma phenotype switching, and resistance to treatment. Cancers 2020, 12, 2158. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable cancer target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Kalinski, P.; Talmadge, J.E. Tumor immuno-environment in cancer progression and therapy. Adv. Exp. Med. Biol. 2017, 1036, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Woo, S.R.; Zha, Y.; Spaapen, R.; Zheng, Y.; Corrales, L.; Spranger, S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 268–276. [Google Scholar] [CrossRef]
- Gravitz, L. Cancer immunotherapy. Nature 2013, 504, S1. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Vasilevska, J.; De Souza, G.A.; Stensland, M.; Skrastina, D.; Zhulenvovs, D.; Paplausks, R.; Kurena, B.; Kozlovska, T.; Zajakina, A. Comparative protein profiling of B16 mouse melanoma cells susceptible and non-susceptible to alphavirus infection: Effect of the tumor microenvironment. Cancer Biol. Ther. 2016, 17, 1035–1050. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Herlyn, M. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 2008, 10, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Figenschau, S.L.; Knutsen, E.; Urbarova, I.; Fenton, C.; Elston, B.; Perander, M.; Mortensen, E.S.; Fenton, K.A. ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Sci. Rep. 2018, 8, 11720. [Google Scholar] [CrossRef] [Green Version]
- Joiner, J.B.; Pylayeva-Gupta, Y.; Dayton, P.A. Focused ultrasound for immunomodulation of the tumor microenvironment. J. Immunol. 2020, 205, 2327–2341. [Google Scholar] [CrossRef] [PubMed]
- Sheybani, N.D.; Price, R.J. Perspectives on recent progress in focused ultrasound immunotherapy. Theranostics 2019, 9, 7749–7758. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Van den Eynde, M.; Bindea, G.; Church, S.E.; Vasaturo, A.; Fredriksen, T.; Lafontaine, L.; Haicheur, N.; Marliot, F.; Debetancourt, D.; et al. Comprehensive intrametastatic immune quantification and major impact of immunoscore on survival. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burks, S.R.; Ziadloo, A.; Kim, S.J.; Nguyen, B.A.; Frank, J.A. Noninvasive pulsed focused ultrasound allows spatiotemporal control of targeted homing for multiple stem cell types in murine skeletal muscle and the magnitude of cell homing can be increased through repeated applications. Stem Cells 2013, 31, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorsung, R.M.; Rosenblatt, R.B.; Cohen, G.; Frank, J.A.; Burks, S.R. Acoustic radiation or cavitation forces from therapeutic ultrasound generate prostaglandins and increase mesenchymal stromal cell homing to murine muscle. Front. Bioeng. Biotechnol. 2020, 8, 870. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, R.B.; Frank, J.A.; Burks, S.R. Cytosolic Ca2+ transients during pulsed focused ultrasound generate reactive oxygen species and cause DNA damage in tumor cells. Theranostics 2021, 11, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Lin, J.; Sun, X.; Ding, Q. Exosomes derived from low-intensity pulsed ultrasound-treated dendritic cells suppress tumor necrosis factor-induced endothelial inflammation. J. Ultrasound Med. 2019, 38, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Sheybani, N.D.; Batts, A.J.; Mathew, A.S.; Thim, E.A.; Price, R.J. Focused ultrasound hyperthermia augments release of glioma-derived extracellular vesicles with differential immunomodulatory capacity. Theranostics 2020, 10, 7436–7447. [Google Scholar] [CrossRef]
- Bhatta, B.; Cooks, T. Reshaping the tumor microenvironment: Extracellular vesicles as messengers of cancer cells. Carcinogenesis 2020, 41, 1461–1470. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, G.; Chandran, P.; Lorsung, R.M.; Aydin, O.; Tomlinson, L.E.; Rosenblatt, R.B.; Burks, S.R.; Frank, J.A. Pulsed-Focused Ultrasound Slows B16 Melanoma and 4T1 Breast Tumor Growth through Differential Tumor Microenvironmental Changes. Cancers 2021, 13, 1546. https://doi.org/10.3390/cancers13071546
Cohen G, Chandran P, Lorsung RM, Aydin O, Tomlinson LE, Rosenblatt RB, Burks SR, Frank JA. Pulsed-Focused Ultrasound Slows B16 Melanoma and 4T1 Breast Tumor Growth through Differential Tumor Microenvironmental Changes. Cancers. 2021; 13(7):1546. https://doi.org/10.3390/cancers13071546
Chicago/Turabian StyleCohen, Gadi, Parwathy Chandran, Rebecca M. Lorsung, Omer Aydin, Lauren E. Tomlinson, Robert B. Rosenblatt, Scott R. Burks, and Joseph A. Frank. 2021. "Pulsed-Focused Ultrasound Slows B16 Melanoma and 4T1 Breast Tumor Growth through Differential Tumor Microenvironmental Changes" Cancers 13, no. 7: 1546. https://doi.org/10.3390/cancers13071546
APA StyleCohen, G., Chandran, P., Lorsung, R. M., Aydin, O., Tomlinson, L. E., Rosenblatt, R. B., Burks, S. R., & Frank, J. A. (2021). Pulsed-Focused Ultrasound Slows B16 Melanoma and 4T1 Breast Tumor Growth through Differential Tumor Microenvironmental Changes. Cancers, 13(7), 1546. https://doi.org/10.3390/cancers13071546