
A Tale of Ice and Fire: The Dual Role for 17β-Estradiol in Balancing DNA Damage and Genome Integrity


 ,
, 

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
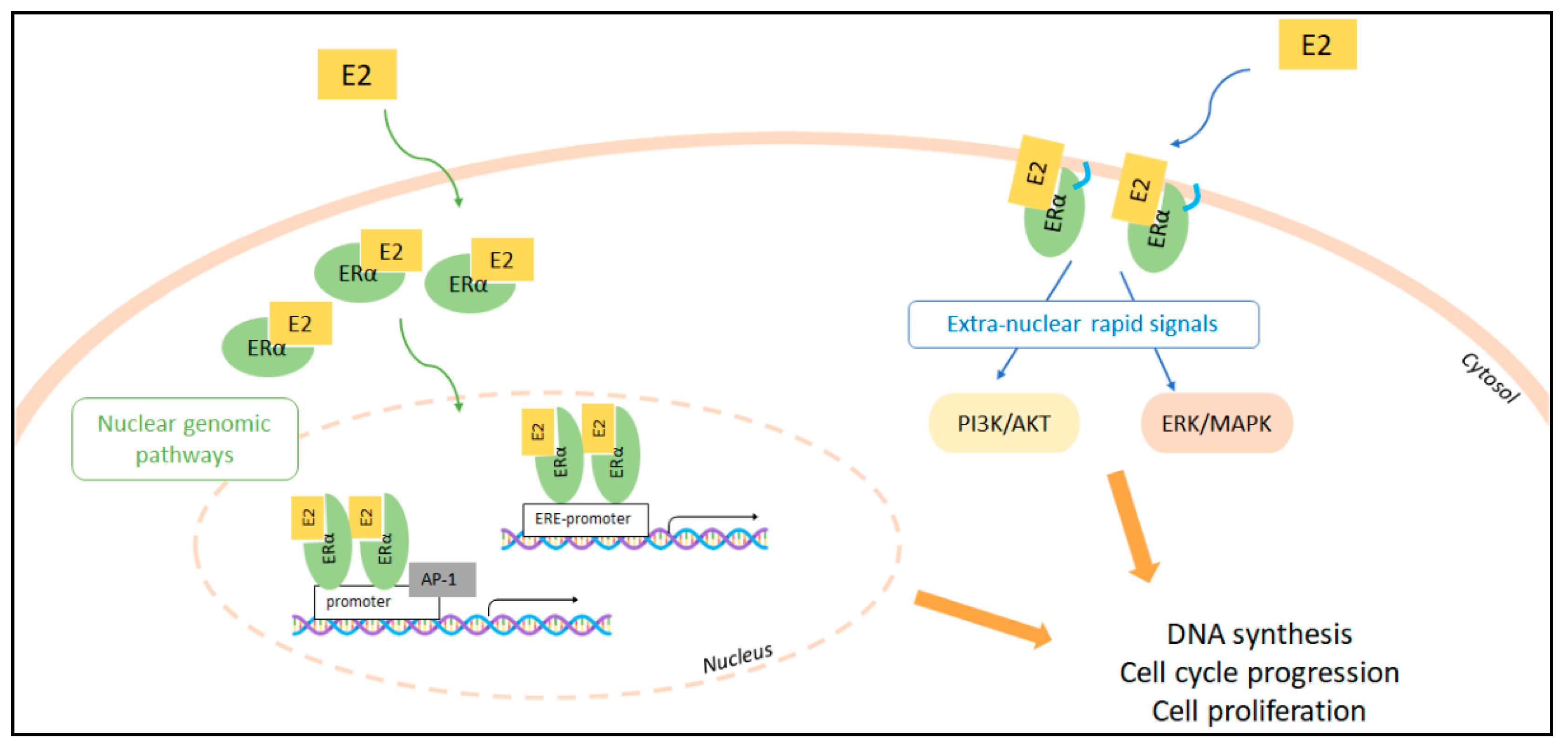
2. The Molecular Pathways of E2:ERα Signaling to Cell Proliferation
Context-Dependent Effects of E2
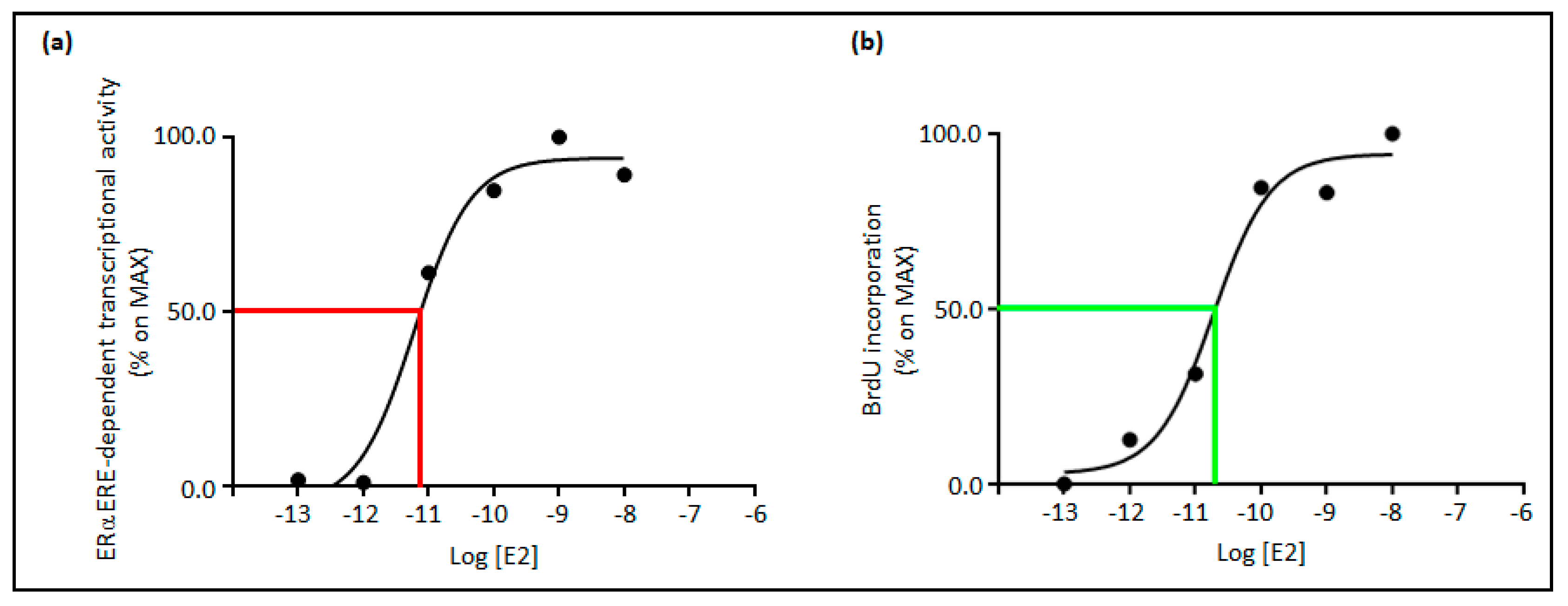
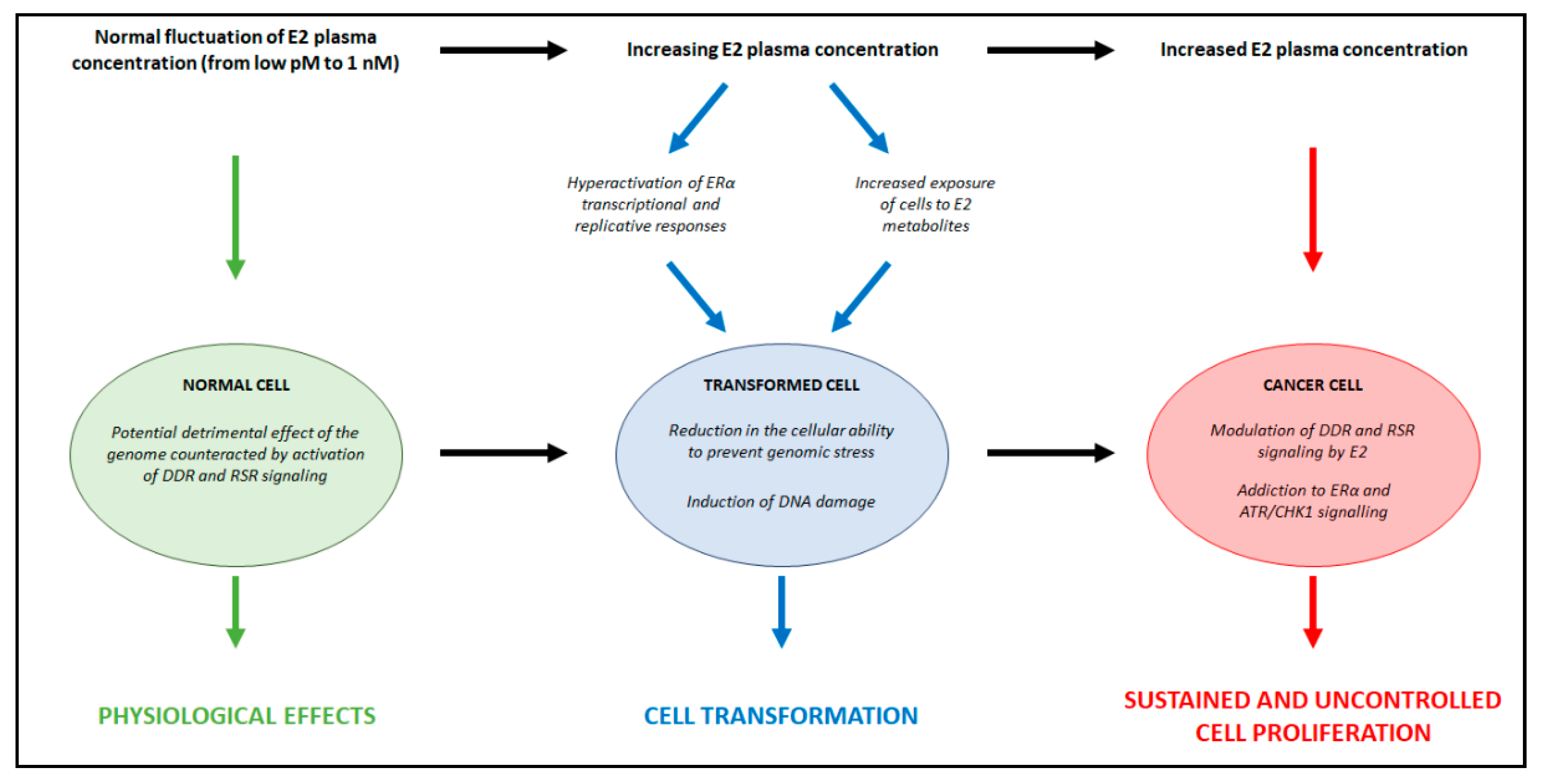
3. Relationships between E2 Concentrations and E2:ERα Signaling to Cell Proliferation
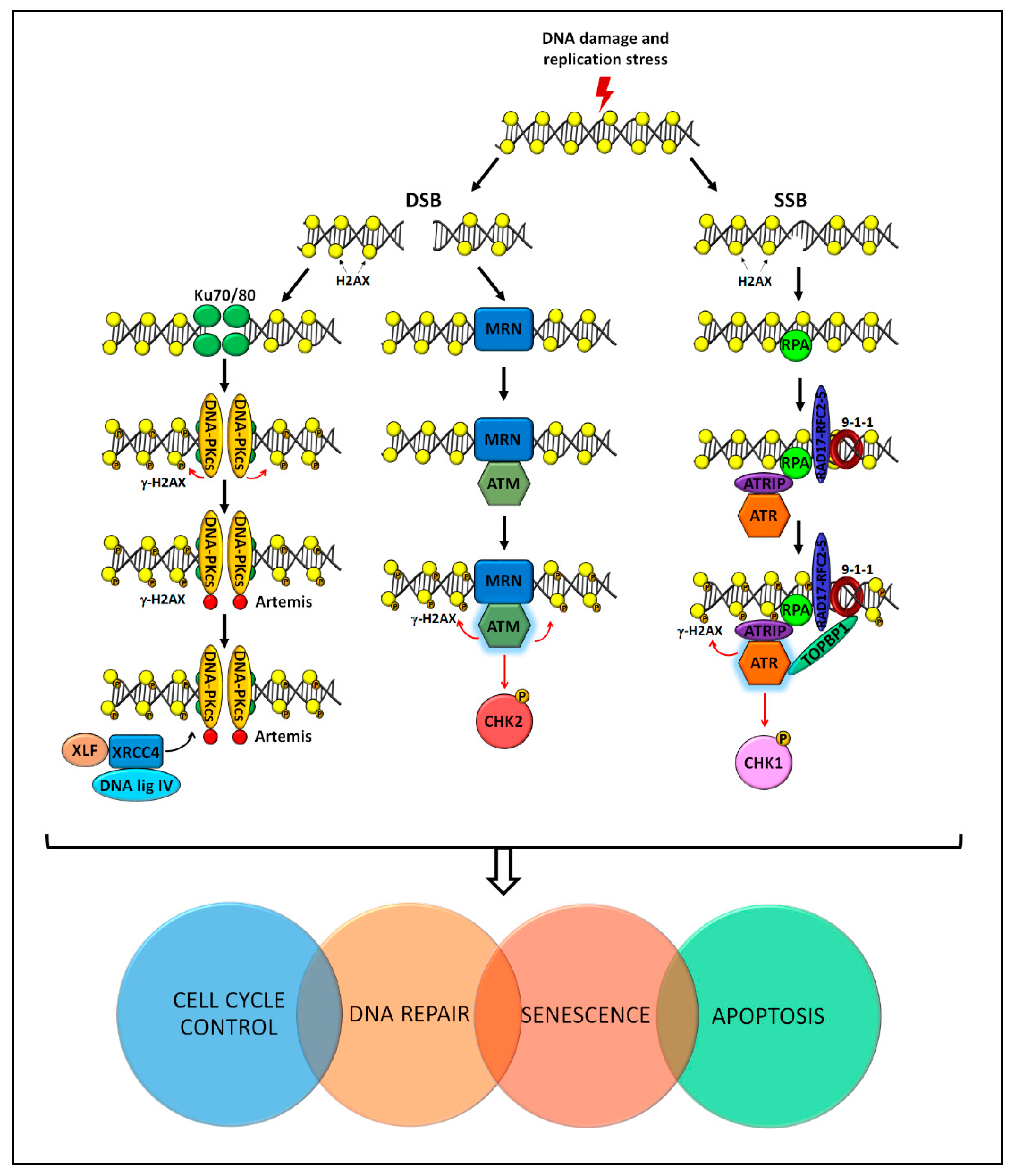
4. Molecular Pathways for Genome Stability Maintenance
5. The Interplay among E2, E2:ERα Signaling, and Genome Stability Maintenance
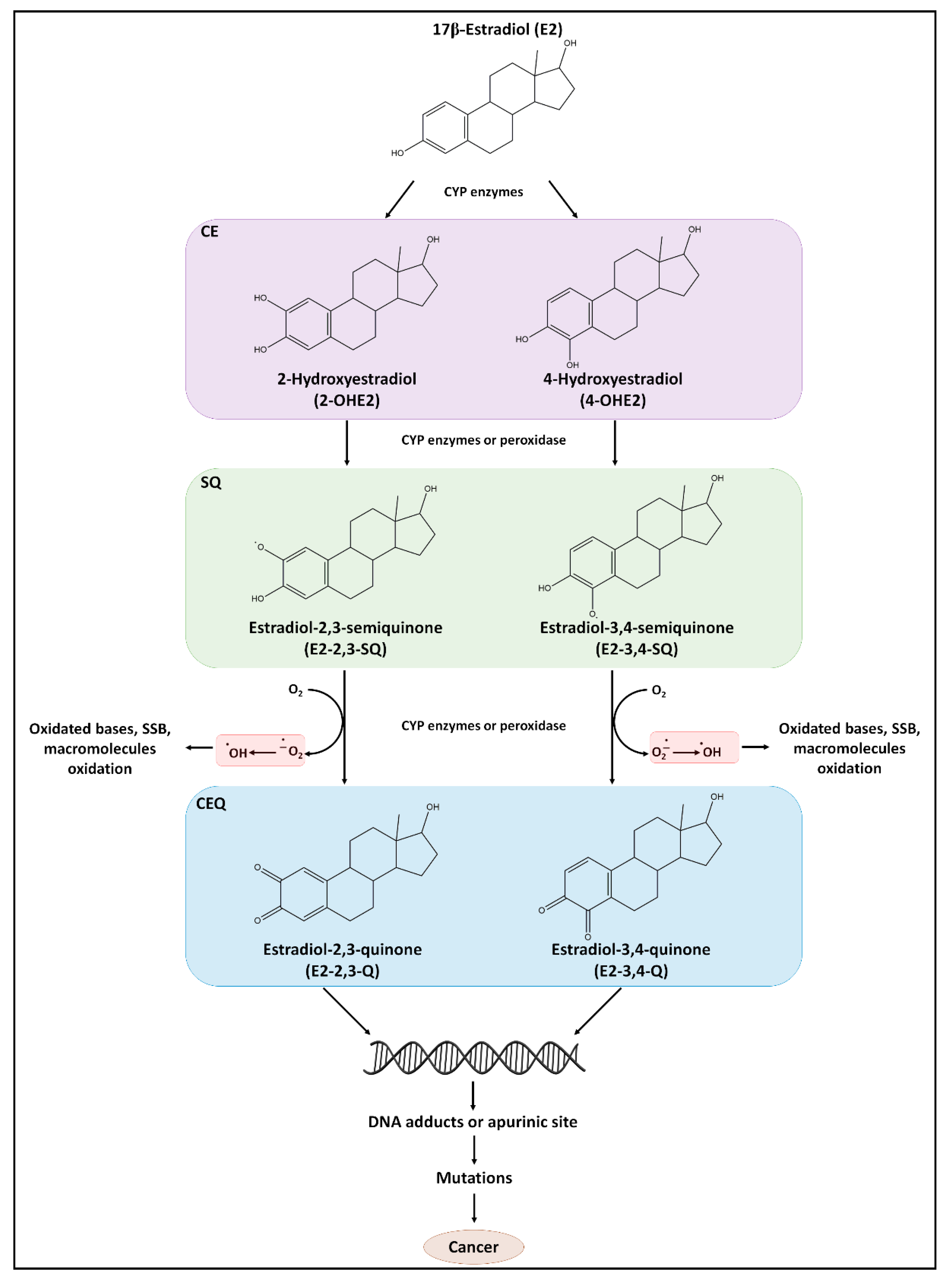
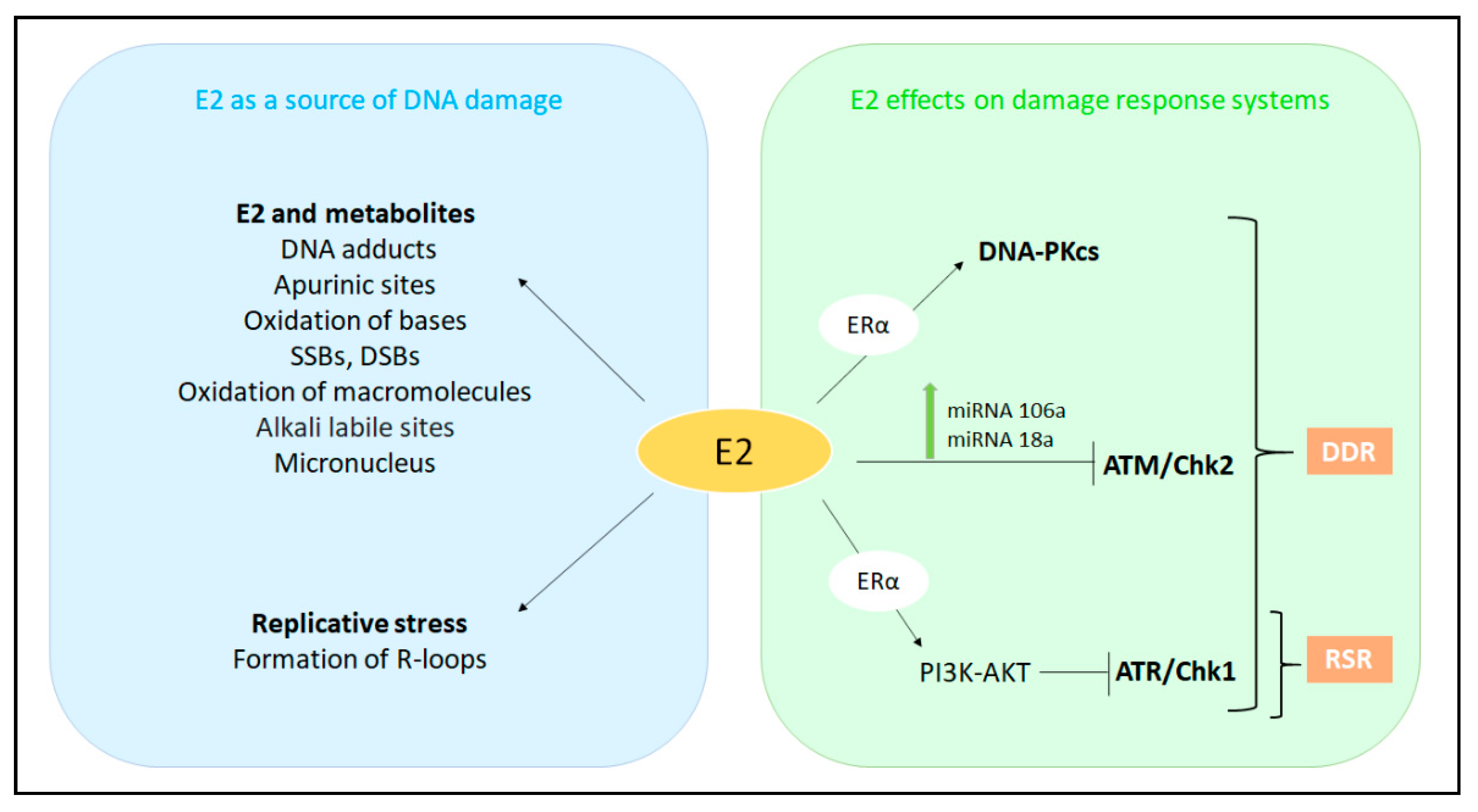
5.1. E2 as a Source of DNA Damage
5.1.1. E2 as a Direct Carcinogen
5.1.2. E2 as a Source of Replicative Stress
5.2. The E2-Dependent Regulation of DDR and RSR Signaling
5.2.1. The E2-Dependent Control of the DNA Damage Response Signaling
5.2.2. The E2-Dependent Control of the Replicative Stress Response Signaling
5.3. Essential Functional Role of DDR and RSR Signaling in the Regulation of E2:ERα-Dependent Cell Proliferation
DDR and RSR Pathways in ERα-Positive BC
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Marino, M. The Effects of 17beta-estradiol in Cancer are Mediated by Estrogen Receptor Signaling at the Plasma Membrane. Front. Physiol. 2011, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Key, T.; Appleby, P.; Barnes, I.; Reeves, G.; Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002, 94, 606–616. [Google Scholar] [CrossRef]
- Farhat, G.N.; Cummings, S.R.; Chlebowski, R.T.; Parimi, N.; Cauley, J.A.; Rohan, T.E.; Huang, A.J.; Vitolins, M.; Hubbell, F.A.; Manson, J.E.; et al. Sex hormone levels and risks of estrogen receptor-negative and estrogen receptor-positive breast cancers. J. Natl. Cancer Inst. 2011, 103, 562–570. [Google Scholar] [CrossRef]
- Eliassen, A.H.; Missmer, S.A.; Tworoger, S.S.; Spiegelman, D.; Barbieri, R.L.; Dowsett, M.; Hankinson, S.E. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J. Natl. Cancer Inst. 2006, 98, 1406–1415. [Google Scholar] [CrossRef]
- Caldon, C.E. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front. Oncol. 2014, 4, 106. [Google Scholar] [CrossRef]
- Liehr, J.G. Is estradiol a genotoxic mutagenic carcinogen? Endocr. Rev. 2000, 21, 40–54. [Google Scholar] [CrossRef]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.M.; Lees-Miller, S.P. Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011, 32, 279–285. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Bantele, S.C.S.; Pfander, B. Quantitative mechanisms of DNA damage sensing and signaling. Curr. Genet. 2020, 66, 59–62. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Kawaguchi, K.; Toi, M. DNA damage repair functions and targeted treatment in breast cancer. Breast Cancer 2020, 27, 355–362. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016, 5. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor alpha membrane localization: Regulation by 17 beta-estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17beta-Estradiol-Induced Estrogen Receptor-alpha Degradation and Transcriptional Activity. Mol. Endocrinol. 2012, 26, 762–774. [Google Scholar] [CrossRef]
- Totta, P.; Busonero, C.; Leone, S.; Marino, M.; Acconcia, F. Dynamin II is required for 17beta-estradiol signaling and autophagy-based ERalpha degradation. Sci. Rep. 2016, 6, 23727. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Lewis, M.; Hammes, S.; Levin, E.R. Membrane-localized estrogen receptor alpha is required for normal organ development and function. Dev. Cell 2014. [Google Scholar] [CrossRef]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef]
- Doisneau-Sixou, S.F.; Sergio, C.M.; Carroll, J.S.; Hui, R.; Musgrove, E.A.; Sutherland, R.L. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 2003, 10, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E.; Daly, R.J.; Sutherland, R.L.; Musgrove, E.A. Cell cycle control in breast cancer cells. J. Cell Biochem. 2006, 97, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Gustafsson, J.A. Estrogen receptors: Therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 2011, 89, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Gigantino, V.; Salvati, A.; Giurato, G.; Palumbo, D.; Strianese, O.; Rizzo, F.; Tarallo, R.; Nyman, T.A.; Weisz, A.; Nassa, G. Identification of Antiestrogen-Bound Estrogen Receptor alpha Interactomes in Hormone-Responsive Human Breast Cancer Cell Nuclei. Proteomics 2020, 20, e2000135. [Google Scholar] [CrossRef]
- Nassa, G.; Tarallo, R.; Guzzi, P.H.; Ferraro, L.; Cirillo, F.; Ravo, M.; Nola, E.; Baumann, M.; Nyman, T.A.; Cannataro, M.; et al. Comparative analysis of nuclear estrogen receptor alpha and beta interactomes in breast cancer cells. Mol. Biosyst. 2011, 7, 667–676. [Google Scholar] [CrossRef]
- Tarallo, R.; Bamundo, A.; Nassa, G.; Nola, E.; Paris, O.; Ambrosino, C.; Facchiano, A.; Baumann, M.; Nyman, T.A.; Weisz, A. Identification of proteins associated with ligand-activated estrogen receptor alpha in human breast cancer cell nuclei by tandem affinity purification and nano LC-MS/MS. Proteomics 2011, 11, 172–179. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Schwabe, J.W.; Chapman, L.; Finch, J.T.; Rhodes, D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: How receptors discriminate between their response elements. Cell 1993, 75, 567–578. [Google Scholar] [CrossRef]
- Bosso, G.; Cipressa, F.; Moroni, M.L.; Pennisi, R.; Albanesi, J.; Brandi, V.; Cugusi, S.; Renda, F.; Ciapponi, L.; Polticelli, F.; et al. NBS1 interacts with HP1 to ensure genome integrity. Cell Death Dis. 2019, 10, 951. [Google Scholar] [CrossRef]
- Dan, P.; Cheung, J.C.; Scriven, D.R.; Moore, E.D. Epitope-dependent localization of estrogen receptor-alpha, but not -beta, in en face arterial endothelium. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1295–H1306. [Google Scholar] [CrossRef]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef]
- van Hoorn, W.P. Identification of a second binding site in the estrogen receptor. J. Med. Chem. 2002, 45, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chirgadze, N.Y.; Briggs, S.L.; Khan, S.; Jensen, E.V.; Burris, T.P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2006, 103, 9908–9911. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Schwarz, I.; Scheld, K.; Sudhop, T.; Berthold, H.K.; von Bergmann, K.; van der Ven, H.; Stehle, P. Physiologic fluctuations of serum estradiol levels influence biochemical markers of bone resorption in young women. J. Clin. Endocrinol. Metab. 2000, 85, 95–101. [Google Scholar] [CrossRef]
- Fontaine, C.; Buscato, M.; Vinel, A.; Giton, F.; Raymond-Letron, I.; Kim, S.H.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Gourdy, P.; Milon, A.; et al. The tissue-specific effects of different 17beta-estradiol doses reveal the key sensitizing role of AF1 domain in ERalpha activity. Mol. Cell Endocrinol. 2020, 505, 110741. [Google Scholar] [CrossRef]
- Metivier, R.; Penot, G.; Hubner, M.R.; Reid, G.; Brand, H.; Kos, M.; Gannon, F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef]
- Reid, G.; Hubner, M.R.; Metivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 2003, 11, 695–707. [Google Scholar] [CrossRef]
- Welboren, W.-J.; Stunnenberg, H. ChIP-Seq Profiling of Estrogen Receptor Alpha Binding Sites Using the Illumina Genome Analyzer; Application Note: Sequencing; Illumina: San Diego, CA, USA, 2010; Available online: https://webcache.googleusercontent.com/search?q=cache:Q2ZB441UfzYJ:https://www.illumina.com/documents/products/appnotes/appnote_chip_sequence_estrogen_receptor_alpha_binding.pdf+&cd=5&hl=it&ct=clnk&gl=it (accessed on 29 March 2021).
- Liu, Z.; Merkurjev, D.; Yang, F.; Li, W.; Oh, S.; Friedman, M.J.; Song, X.; Zhang, F.; Ma, Q.; Ohgi, K.A.; et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 2014, 159, 358–373. [Google Scholar] [CrossRef]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Cipolletti, M.; Leone, S.; Bartoloni, S.; Busonero, C.; Acconcia, F. Real-time measurement of E2: ERalpha transcriptional activity in living cells. J. Cell Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Acconcia, F.; Bresciani, F.; Weisz, A.; Trentalance, A. Distinct nongenomic signal transduction pathways controlled by 17beta-estradiol regulate DNA synthesis and cyclin D(1) gene transcription in HepG2 cells. Mol. Biol. Cell 2002, 13, 3720–3729. [Google Scholar] [CrossRef]
- Marino, M.; Acconcia, F.; Trentalance, A. Biphasic estradiol-induced AKT phosphorylation is modulated by PTEN via MAP kinase in HepG2 cells. Mol. Biol. Cell 2003, 14, 2583–2591. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Munoz, S.; Mendez, J. DNA replication stress: From molecular mechanisms to human disease. Chromosoma 2017, 126, 1–15. [Google Scholar] [CrossRef]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef]
- Suzuki, K.; Kodama, S.; Watanabe, M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J. Biol. Chem. 1999, 274, 25571–25575. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007, 26, 7749–7758. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Byun, T.; Yee, M.C.; Hekmat-Nejad, M.; Cimprich, K.A. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002, 16, 2327–2332. [Google Scholar] [CrossRef]
- Dart, D.A.; Adams, K.E.; Akerman, I.; Lakin, N.D. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem. 2004, 279, 16433–16440. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Bronk, G.; Kondev, J.; Haber, J.E. Yeast ATM and ATR kinases use different mechanisms to spread histone H2A phosphorylation around a DNA double-strand break. Proc. Natl. Acad. Sci. USA 2020, 117, 21354–21363. [Google Scholar] [CrossRef]
- Damia, G. Targeting DNA-PK in cancer. Mutat. Res. 2020, 821, 111692. [Google Scholar] [CrossRef]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Van, H.T.; Santos, M.A. Histone modifications and the DNA double-strand break response. Cell Cycle 2018, 17, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Rona, G.; Pagano, M. Mixed ubiquitin chains regulate DNA repair. Genes Dev. 2019, 33, 1615–1616. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.H.; Chen, S.H.; Yu, X. Poly-ADP ribosylation in DNA damage response and cancer therapy. Mutat. Res. 2019, 780, 82–91. [Google Scholar] [CrossRef]
- Xie, M.; Yu, J.; Ge, S.; Huang, J.; Fan, X. SUMOylation homeostasis in tumorigenesis. Cancer Lett. 2020, 469, 301–309. [Google Scholar] [CrossRef]
- Yager, J.D. Mechanisms of estrogen carcinogenesis: The role of E2/E1-quinone metabolites suggests new approaches to preventive intervention—A review. Steroids 2015, 99, 56–60. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef]
- Huang, J.; Sun, J.; Chen, Y.; Song, Y.; Dong, L.; Zhan, Q.; Zhang, R.; Abliz, Z. Analysis of multiplex endogenous estrogen metabolites in human urine using ultra-fast liquid chromatography-tandem mass spectrometry: A case study for breast cancer. Anal. Chim. Acta 2012, 711, 60–68. [Google Scholar] [CrossRef]
- Rogan, E.G.; Badawi, A.F.; Devanesan, P.D.; Meza, J.L.; Edney, J.A.; West, W.W.; Higginbotham, S.M.; Cavalieri, E.L. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis 2003, 24, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Stack, D.E.; Devanesan, P.D.; Todorovic, R.; Dwivedy, I.; Higginbotham, S.; Johansson, S.L.; Patil, K.D.; Gross, M.L.; Gooden, J.K.; et al. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA 1997, 94, 10937–10942. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Kohli, E.; Saeed, M.; Rogan, E.; Cavalieri, E. The greater reactivity of estradiol-3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: Implications for tumor-initiating activity. Chem. Res. Toxicol. 2006, 19, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Rogan, E.G. Depurinating estrogen-DNA adducts, generators of cancer initiation: Their minimization leads to cancer prevention. Clin. Transl. Med. 2016, 5, 12. [Google Scholar] [CrossRef]
- Mannisto, P.T.; Kaakkola, S. Catechol-O-methyltransferase (COMT): Biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol. Rev. 1999, 51, 593–628. [Google Scholar]
- Zahid, M.; Saeed, M.; Lu, F.; Gaikwad, N.; Rogan, E.; Cavalieri, E. Inhibition of catechol-O-methyltransferase increases estrogen-DNA adduct formation. Free Radic. Biol. Med. 2007, 43, 1534–1540. [Google Scholar] [CrossRef]
- Wu, Q.; Odwin-Dacosta, S.; Cao, S.; Yager, J.D.; Tang, W.Y. Estrogen down regulates COMT transcription via promoter DNA methylation in human breast cancer cells. Toxicol. Appl. Pharmacol. 2019, 367, 12–22. [Google Scholar] [CrossRef]
- Hachey, D.L.; Dawling, S.; Roodi, N.; Parl, F.F. Sequential action of phase I and II enzymes cytochrome p450 1B1 and glutathione S-transferase P1 in mammary estrogen metabolism. Cancer Res. 2003, 63, 8492–8499. [Google Scholar] [PubMed]
- Lu, F.; Zahid, M.; Wang, C.; Saeed, M.; Cavalieri, E.L.; Rogan, E.G. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prev. Res. 2008, 1, 135–145. [Google Scholar] [CrossRef]
- Zahid, M.; Gaikwad, N.W.; Ali, M.F.; Lu, F.; Saeed, M.; Yang, L.; Rogan, E.G.; Cavalieri, E.L. Prevention of estrogen-DNA adduct formation in MCF-10F cells by resveratrol. Free Radic. Biol. Med. 2008, 45, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Liehr, J.G. Induction by estrogens of lipid peroxidation and lipid peroxide-derived malonaldehyde-DNA adducts in male Syrian hamsters: Role of lipid peroxidation in estrogen-induced kidney carcinogenesis. Carcinogenesis 1995, 16, 1941–1945. [Google Scholar] [CrossRef] [PubMed]
- Mobley, J.A.; Brueggemeier, R.W. Estrogen receptor-mediated regulation of oxidative stress and DNA damage in breast cancer. Carcinogenesis 2004, 25, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Pinteric, M.; Podgorski, I.I.; Hadzija, M.P.; Filic, V.; Paradzik, M.; Proust, B.L.J.; Dekanic, A.; Ciganek, I.; Plese, D.; Marcinko, D.; et al. Sirt3 Exerts Its Tumor-Suppressive Role by Increasing p53 and Attenuating Response to Estrogen in MCF-7 Cells. Antioxidants 2020, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, N.; Butterworth, M.; Kortenkamp, A. Detection of DNA strand breaks and oxidized DNA bases at the single-cell level resulting from exposure to estradiol and hydroxylated metabolites. Environ. Mol. Mutagen. 2005, 45, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Yared, E.; McMillan, T.J.; Martin, F.L. Genotoxic effects of oestrogens in breast cells detected by the micronucleus assay and the Comet assay. Mutagenesis 2002, 17, 345–352. [Google Scholar] [CrossRef]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Callen Moreu, E.; et al. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Tsuda, M.; Bunch, H.; Sasanuma, H.; Austin, C.; Takeda, S. Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes 2019, 10, 868. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.A.; Weiss, N.S.; Freeman, R.J.; Dightman, D.A.; Thornton, P.J.; Houck, J.R.; Voigt, L.F.; Rossing, M.A.; Schwartz, S.M.; Chen, C. Genetic factors in catechol estrogen metabolism in relation to the risk of endometrial cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 357–366. [Google Scholar] [CrossRef]
- Wang, Q.; Li, H.; Tao, P.; Wang, Y.P.; Yuan, P.; Yang, C.X.; Li, J.Y.; Yang, F.; Lee, H.; Huang, Y. Soy isoflavones, CYP1A1, CYP1B1, and COMT polymorphisms, and breast cancer: A case-control study in southwestern China. DNA Cell Biol. 2011, 30, 585–595. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Madak-Erdogan, Z.; Li, J.; Cheng, J.; Greenlief, C.M.; Helferich, W.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Transcriptomic analysis identifies gene networks regulated by estrogen receptor alpha (ERalpha) and ERbeta that control distinct effects of different botanical estrogens. Nucl. Recept. Signal. 2014, 12, e001. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef]
- Dvir, A.; Stein, L.Y.; Calore, B.L.; Dynan, W.S. Purification and characterization of a template-associated protein kinase that phosphorylates RNA polymerase II. J. Biol. Chem. 1993, 268, 10440–10447. [Google Scholar] [CrossRef]
- Anderson, C.W.; Lees-Miller, S.P. The nuclear serine/threonine protein kinase DNA-PK. Crit. Rev. Eukaryot Gene Expr. 1992, 2, 283–314. [Google Scholar] [PubMed]
- Medunjanin, S.; Weinert, S.; Schmeisser, A.; Mayer, D.; Braun-Dullaeus, R.C. Interaction of the double-strand break repair kinase DNA-PK and estrogen receptor-alpha. Mol. Biol. Cell 2010, 21, 1620–1628. [Google Scholar] [CrossRef]
- Medunjanin, S.; Weinert, S.; Poitz, D.; Schmeisser, A.; Strasser, R.H.; Braun-Dullaeus, R.C. Transcriptional activation of DNA-dependent protein kinase catalytic subunit gene expression by oestrogen receptor-alpha. EMBO Rep. 2010, 11, 208–213. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636, Erratum in 2017, 18, 783, doi:10.1038/nrm.2017.116. [Google Scholar] [CrossRef]
- Kumagai, A.; Dunphy, W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar] [CrossRef]
- Chini, C.C.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Kacmaz, K.; Chastain, P.D.; Qu, P.P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Gutierrez-Martinez, P.; Soria, R.; Fernandez-Capetillo, O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008, 22, 297–302. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montana, M.F.; Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Fernandez-Capetillo, O. The ATR barrier to replication-born DNA damage. DNA Repair. 2010, 9, 1249–1255. [Google Scholar] [CrossRef]
- Bartek, J.; Mistrik, M.; Bartkova, J. Thresholds of replication stress signaling in cancer development and treatment. Nat. Struct. Mol. Biol. 2012, 19, 5–7. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Evinger, A.J.; Lee, E.; Levin, E.R. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol. Biol. Cell 2009, 20, 3374–3389. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Lin, C.; Wu, Z.; Gong, H.; Zeng, Y.; Wu, J.; Li, M.; Li, J. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS ONE 2011, 6, e25454. [Google Scholar] [CrossRef]
- Smits, V.A.J.; Cabrera, E.; Freire, R.; Gillespie, D.A. Claspin—Checkpoint adaptor and DNA replication factor. FEBS J. 2019, 286, 441–455. [Google Scholar] [CrossRef]
- Cho, W.H.; Kang, Y.H.; An, Y.Y.; Tappin, I.; Hurwitz, J.; Lee, J.K. Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc. Natl. Acad. Sci. USA 2013, 110, 2523–2527. [Google Scholar] [CrossRef]
- Bianco, J.N.; Bergoglio, V.; Lin, Y.L.; Pillaire, M.J.; Schmitz, A.L.; Gilhodes, J.; Lusque, A.; Mazieres, J.; Lacroix-Triki, M.; Roumeliotis, T.I.; et al. Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat. Commun. 2019, 10, 910. [Google Scholar] [CrossRef] [PubMed]
- Magne Nde, C.B.; Casas Gimeno, G.; Docanto, M.; Knower, K.C.; Young, M.J.; Buehn, J.; Sayed, E.; Clyne, C.D. Timeless Is a Novel Estrogen Receptor Co-activator Involved in Multiple Signaling Pathways in MCF-7 Cells. J. Mol. Biol. 2018, 430, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Tsimaratou, K.; Kletsas, D.; Kastrinakis, N.G.; Tsantoulis, P.K.; Evangelou, K.; Sideridou, M.; Liontos, M.; Poulias, I.; Venere, M.; Salmas, M.; et al. Evaluation of claspin as a proliferation marker in human cancer and normal tissues. J. Pathol. 2007, 211, 331–339. [Google Scholar] [CrossRef]
- Mao, Y.; Fu, A.; Leaderer, D.; Zheng, T.; Chen, K.; Zhu, Y. Potential cancer-related role of circadian gene TIMELESS suggested by expression profiling and in vitro analyses. BMC Cancer 2013, 13, 498. [Google Scholar] [CrossRef] [PubMed]
- Baldeyron, C.; Brisson, A.; Tesson, B.; Nemati, F.; Koundrioukoff, S.; Saliba, E.; De Koning, L.; Martel, E.; Ye, M.; Rigaill, G.; et al. TIPIN depletion leads to apoptosis in breast cancer cells. Mol. Oncol. 2015, 9, 1580–1598. [Google Scholar] [CrossRef]
- Tozlu-Kara, S.; Roux, V.; Andrieu, C.; Vendrell, J.; Vacher, S.; Lazar, V.; Spyratos, F.; Tubiana-Hulin, M.; Cohen, P.; Dessen, P.; et al. Oligonucleotide microarray analysis of estrogen receptor alpha-positive postmenopausal breast carcinomas: Identification of HRPAP20 and TIMELESS as outstanding candidate markers to predict the response to tamoxifen. J. Mol. Endocrinol. 2007, 39, 305–318. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Billon, P.; Bryant, E.E.; Joseph, S.A.; Nambiar, T.S.; Hayward, S.B.; Rothstein, R.; Ciccia, A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell 2017, 67, 1068–1079.e4. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Gigantino, V.; Nassa, G.; Mirici Cappa, V.; Ventola, G.M.; Cracas, D.G.C.; Mastrocinque, R.; Rizzo, F.; Tarallo, R.; Weisz, A.; et al. Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 4068. [Google Scholar] [CrossRef] [PubMed]
- Kitao, H.; Iimori, M.; Kataoka, Y.; Wakasa, T.; Tokunaga, E.; Saeki, H.; Oki, E.; Maehara, Y. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018, 109, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yang, C.; Qian, X.; Lei, T.; Li, Y.; Shen, H.; Fu, L.; Xu, B. Estrogen receptor alpha regulates ATM Expression through miRNAs in breast cancer. Clin. Cancer Res. 2013, 19, 4994–5002. [Google Scholar] [CrossRef]
- Ayres, S.; Abplanalp, W.; Liu, J.H.; Subbiah, M.T. Mechanisms involved in the protective effect of estradiol-17beta on lipid peroxidation and DNA damage. Am. J. Physiol. 1998, 274, E1002–E1008. [Google Scholar] [CrossRef]
- Stepniak, J.; Karbownik-Lewinska, M. 17beta-estradiol prevents experimentally-induced oxidative damage to membrane lipids and nuclear DNA in porcine ovary. Syst. Biol. Reprod. Med. 2016, 62, 17–21. [Google Scholar] [CrossRef]
- Savage, K.I.; Matchett, K.B.; Barros, E.M.; Cooper, K.M.; Irwin, G.W.; Gorski, J.J.; Orr, K.S.; Vohhodina, J.; Kavanagh, J.N.; Madden, A.F.; et al. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res. 2014, 74, 2773–2784. [Google Scholar] [CrossRef]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pescatori, S.; Berardinelli, F.; Albanesi, J.; Ascenzi, P.; Marino, M.; Antoccia, A.; di Masi, A.; Acconcia, F. A Tale of Ice and Fire: The Dual Role for 17β-Estradiol in Balancing DNA Damage and Genome Integrity. Cancers 2021, 13, 1583. https://doi.org/10.3390/cancers13071583
Pescatori S, Berardinelli F, Albanesi J, Ascenzi P, Marino M, Antoccia A, di Masi A, Acconcia F. A Tale of Ice and Fire: The Dual Role for 17β-Estradiol in Balancing DNA Damage and Genome Integrity. Cancers. 2021; 13(7):1583. https://doi.org/10.3390/cancers13071583
Chicago/Turabian StylePescatori, Sara, Francesco Berardinelli, Jacopo Albanesi, Paolo Ascenzi, Maria Marino, Antonio Antoccia, Alessandra di Masi, and Filippo Acconcia. 2021. "A Tale of Ice and Fire: The Dual Role for 17β-Estradiol in Balancing DNA Damage and Genome Integrity" Cancers 13, no. 7: 1583. https://doi.org/10.3390/cancers13071583
APA StylePescatori, S., Berardinelli, F., Albanesi, J., Ascenzi, P., Marino, M., Antoccia, A., di Masi, A., & Acconcia, F. (2021). A Tale of Ice and Fire: The Dual Role for 17β-Estradiol in Balancing DNA Damage and Genome Integrity. Cancers, 13(7), 1583. https://doi.org/10.3390/cancers13071583