
CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
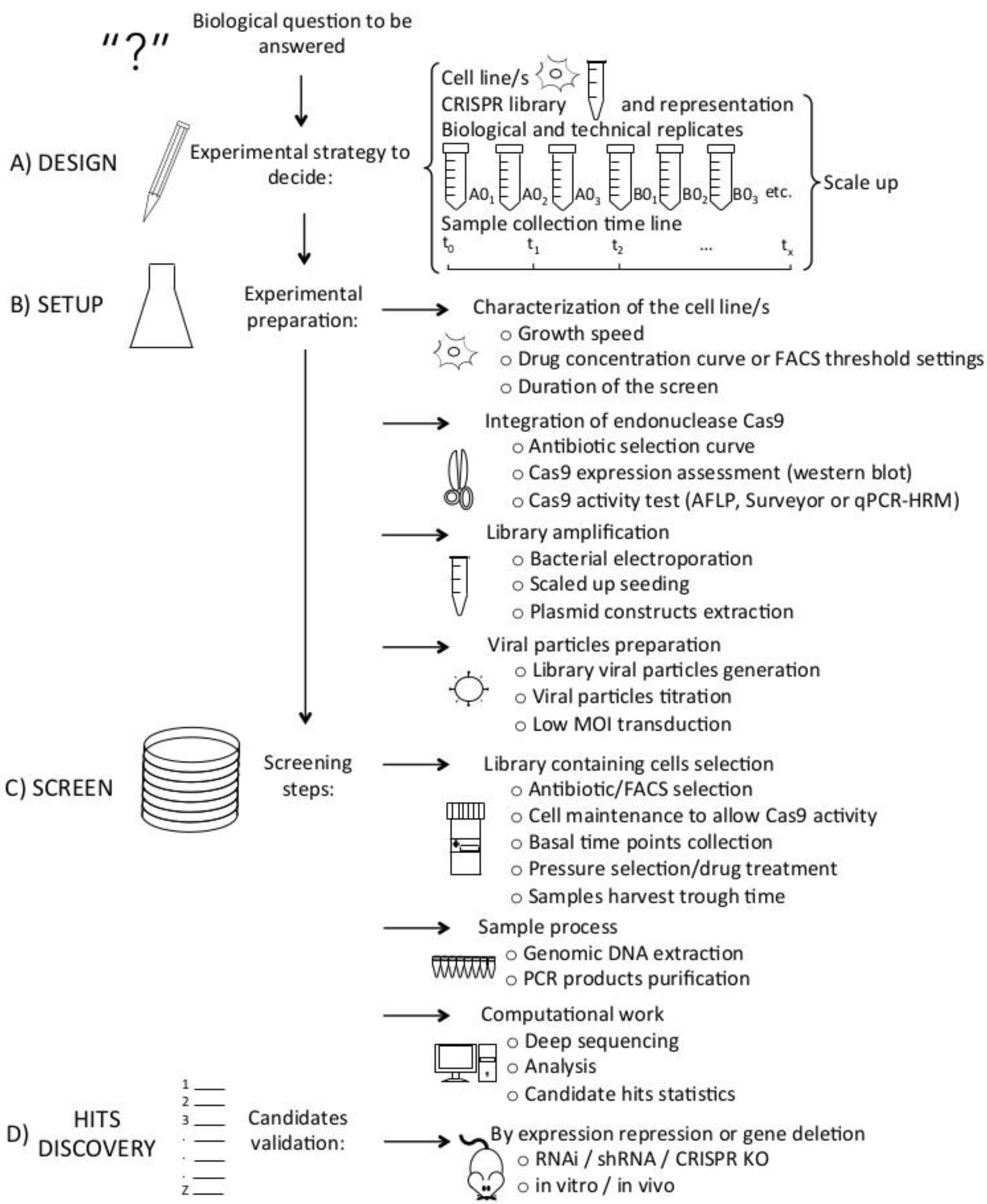
2. CRISPR/Cas9 Screens Steps
2.1. CRISPR Library
2.2. Considerations for Screening in Cancer Cell Lines
2.3. Setting Up the Experimental Protocol
2.4. CRISPR Screen
2.5. Screen Analysis
2.6. Candidate Hits Validation
3. CRISPR/Cas Screens in Cancer
3.1. Synthetic Lethal Screens
3.2. Synthetic Viable Screens
3.3. Novel Drug Targets Screens
3.4. Pooled CRISPR Screens Based on FACS
3.5. 3D Cultured Cancer Models CRISPR Screens
3.6. Ex Vivo and In Vivo CRISPR Screens
3.7. Non-Coding Gene Targets CRISPR Screens
4. Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abascal, F.; Acosta, R.; Addleman, N.J.; Adrian, J.; Afzal, V.; Aken, B.; Akiyama, J.A.; Al Jammal, O.; Amrhein, H.; Anderson, S.M.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020. [Google Scholar] [CrossRef]
- Van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A. CRISPR-Cas immunity in prokaryotes. Nature 2015, 526, 55–61. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Paschon, D.E.; Lussier, S.; Wangzor, T.; Xia, D.F.; Li, P.W.; Hinkley, S.J.; Scarlott, N.A.; Lam, S.C.; Waite, A.J.; Truong, L.N.; et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Voytas, D.F. TAL effectors: Customizable proteins for DNA targeting. Science 2011, 333, 1843–1846. [Google Scholar] [CrossRef]
- Shekar, M.; Venugopal, M.N. Insight into a Transcriptional Adaptor Zinc Finger Encoded by a Putative Protein in the White Spot Syndrome Virus Genome. Interdiscip. Sci. Comput. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; et al. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013, 232, 116636. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Molina, R.; Sofos, N.; Montoya, G. Structural basis of CRISPR-Cas Type III prokaryotic defence systems. Curr. Opin. Struct. Biol. 2020, 65, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014. [Google Scholar] [CrossRef] [PubMed]
- Koike-Yusa, H.; Li, Y.; Tan, E.P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014. [Google Scholar] [CrossRef]
- Hart, T.; Brown, K.R.; Sircoulomb, F.; Rottapel, R.; Moffat, J. Measuring error rates in genomic perturbation screens: Gold standards for human functional genomics. Mol. Syst. Biol. 2014. [Google Scholar] [CrossRef]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Behan, F.M.; Gonçalves, E.; Bhosle, S.G.; Chen, E.; Shepherd, R.; Beaver, C.; Ansari, R.; Pooley, R.; Wilkinson, P.; et al. Unsupervised correction of gene-independent cell responses to CRISPR-Cas9 targeting. BMC Genom. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Malyutina, A.; Pessia, A.; Saarela, J.; Heckman, C.A.; Tang, J. Combined gene essentiality scoring improves the prediction of cancer dependency maps. EBioMedicine 2019. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Leonardo, M.; Zairan, L.; Brar, G.A.; Torres, S.E.; Noam, S.G.; Onn, B.; Whitehead, E.H.; Doudna, J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Horlbeck, M.A.; Gilbert, L.A.; Villalta, J.E.; Adamson, B.; Pak, R.A.; Chen, Y.; Fields, A.P.; Park, C.Y.; Corn, J.E.; Kampmann, M.; et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 2016. [Google Scholar] [CrossRef]
- Davis, E.M.; Sun, Y.; Liu, Y.; Kolekar, P.; Shao, Y.; Szlachta, K.; Mulder, H.L.; Ren, D.; Rice, S.V.; Wang, Z.; et al. SequencErr: Measuring and suppressing sequencer errors in next-generation sequencing data. Genome Biol. 2021. [Google Scholar] [CrossRef]
- Wells, A.; Heckerman, D.; Torkamani, A.; Yin, L.; Sebat, J.; Ren, B.; Telenti, A.; di Iulio, J. Ranking of non-coding pathogenic variants and putative essential regions of the human genome. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Xu, H.; Harrington, W.; Gill, S.; Wang, X.; Vazquez, F.; Root, D.E.; Tsherniak, A.; Hahn, W.C. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, Y.; Zhou, X.; Wright, S.; Hyle, J.; Zhao, L.; An, J.; Zhao, X.; Shao, Y.; Xu, B.; et al. Functional interrogation of HOXA9 regulome in MLLr leukemia via reporter-based CRISPR/Cas9 screen. eLife 2020. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, J.; Cheng, M.; Liao, X.; Peng, S. Review of CRISPR/Cas9 sgRNA Design Tools. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 455–465. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, W.; An, J.H.; Lee, J.H.; Kim, Y.H.; Kim, H.; Oh, Y.; Park, Y.H.; Jin, Y.B.; Jun, B.H.; et al. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment. Nat. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Shou, J. Toward precision CRISPR DNA fragment editing and predictable 3D genome engineering. J. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Sledzinski, P.; Nowaczyk, M.; Olejniczak, M. Computational Tools and Resources Supporting CRISPR-Cas Experiments. Cells 2020, 9, 1288. [Google Scholar] [CrossRef]
- Morgens, D.W.; Wainberg, M.; Boyle, E.A.; Ursu, O.; Araya, C.L.; Kimberly Tsui, C.; Haney, M.S.; Hess, G.T.; Han, K.; Jeng, E.E.; et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Tong, A.H.Y.; Chan, K.; Van Leeuwen, J.; Seetharaman, A.; Aregger, M.; Chandrashekhar, M.; Hustedt, N.; Seth, S.; Noonan, A.; et al. Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3 Genes Genomes Genet. 2017. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009. [Google Scholar] [CrossRef] [Green Version]
- Ferreira da Silva, J.; Salic, S.; Wiedner, M.; Datlinger, P.; Essletzbichler, P.; Hanzl, A.; Superti-Furga, G.; Bock, C.; Winter, G.; Loizou, J.I. Genome-scale CRISPR screens are efficient in non-homologous end-joining deficient cells. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Chan, K.; Tong, A.H.Y.; Brown, K.R.; Mero, P.; Moffat, J. Pooled CRISPR-based genetic screens in Mammalian cells. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Mei, D.Y.; Liu, Q.N.; Qiao, X.H.; Ruan, W.M.; Huang, T.; Cao, G.S. Research of methods to detect genomic mutations induced by CRISPR/Cas systems. J. Biotechnol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Guerriero, M.L.; Corrigan, A.; Bornot, A.; Firth, M.; O’Shea, P.; Ross-Thriepland, D.; Peel, S. Delivering Robust Candidates to the Drug Pipeline through Computational Analysis of Arrayed CRISPR Screens. SLAS Discov. 2020. [Google Scholar] [CrossRef]
- Madsen, R.R.; Semple, R.K. Luminescent peptide tagging enables efficient screening for crispr-mediated knock-in in human induced pluripotent stem cells. Wellcome Open Res. 2019. [Google Scholar] [CrossRef]
- Attayek, P.J.; Waugh, J.P.; Hunsucker, S.A.; Grayeski, P.J.; Sims, C.E.; Armistead, P.M.; Allbritton, N.L. Automated microraft platform to identify and collect non-adherent cells successfully gene-edited with CRISPR-Cas9. Biosens. Bioelectron. 2017. [Google Scholar] [CrossRef] [Green Version]
- Condon, K.J.; Orozco, J.M.; Adelmann, C.H.; Spinelli, J.B.; Van Der Helm, P.W.; Roberts, J.M.; Kunchok, T.; Sabatini, D.M.; Howard, B. Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. bioRxiv 2020. [Google Scholar]
- Cooper, S.E.; Schwartzentruber, J.; Bello, E.; Coomber, E.L.; Bassett, A.R. Screening for functional transcriptional and splicing regulatory variants with GenIE. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Luo, B.; Hiu, W.C.; Subramanian, A.; Sharifnia, T.; Okamoto, M.; Yang, X.; Hinkle, G.; Boehm, J.S.; Beroukhim, R.; Weir, B.A.; et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, R.; Chiang, C.Y.; Tu, B.P.; Yan, S.F.; DeJesus, P.D.; Romero, A.; Bergauer, T.; Orth, A.; Krueger, U.; Zhou, Y.; et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 2007. [Google Scholar] [CrossRef]
- Morgens, D.W.; Deans, R.M.; Li, A.; Bassik, M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014. [Google Scholar] [CrossRef]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.H.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019. [Google Scholar] [CrossRef]
- Winter, J.; Schwering, M.; Pelz, O.; Rauscher, B.; Zhan, T.; Heigwer, F.; Boutros, M. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Winter, J.; Breinig, M.; Heigwer, F.; Brügemann, D.; Leible, S.; Pelz, O.; Zhan, T.; Boutros, M. CaRpools: An R package for exploratory data analysis and documentation of pooled CRISPR/Cas9 screens. Bioinformatics 2016. [Google Scholar] [CrossRef] [Green Version]
- Bodapati, S.; Daley, T.P.; Lin, X.; Zou, J.; Qi, L.S. A benchmark of algorithms for the analysis of pooled CRISPR screens. Genome Biol. 2020, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, M.R.; Kostyrko, K.; Han, K.; Mooney, N.A.; Jeng, E.E.; Spees, K.; Dinh, P.T.; Abbott, K.L.; Gwinn, D.M.; Sweet-Cordero, E.A.; et al. Combined Proteomic and Genetic Interaction Mapping Reveals New RAS Effector Pathways and Susceptibilities. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hinze, L.; Pfirrmann, M.; Karim, S.; Degar, J.; McGuckin, C.; Vinjamur, D.; Sacher, J.; Stevenson, K.E.; Neuberg, D.S.; Orellana, E.; et al. Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias. Cancer Cell 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Zhu, Q.; Liang, J.; Li, Y.; Li, M.; Zhang, Y.; Wang, X.; Zeng, Y.; Jiao, Y. A CRISPR knockout negative screen reveals synergy between CDKs inhibitor and metformin in the treatment of human cancer in vitro and in vivo. Signal. Transduct. Target. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017. [Google Scholar] [CrossRef]
- Li, F.; Ng, W.-L.; Luster, T.A.; Hu, H.; Sviderskiy, V.O.; Dowling, C.M.; Hollinshead, K.E.R.; Zouitine, P.; Zhang, H.; Huang, Q.; et al. Epigenetic CRISPR Screens Identify Npm1 as a Therapeutic Vulnerability in Non–Small Cell Lung Cancer. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Chen, A.; Wen, S.; Liu, F.; Zhang, Z.; Liu, M.; Wu, Y.; He, B.; Yan, M.; Kang, T.; Lam, E.W.F.; et al. CRISPR/Cas9 screening identifies a kinetochore-microtubule dependent mechanism for Aurora-A inhibitor resistance in breast cancer. Cancer Commun. 2021. [Google Scholar] [CrossRef]
- Makhov, P.; Sohn, J.A.; Serebriiskii, I.G.; Fazliyeva, R.; Khazak, V.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. CRISPR/Cas9 genome-wide loss-of-function screening identifies druggable cellular factors involved in sunitinib resistance in renal cell carcinoma. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- Dwane, L.; Behan, F.M.; Gonçalves, E.; Lightfoot, H.; Yang, W.; Van Der Meer, D.; Shepherd, R.; Pignatelli, M.; Iorio, F.; Garnett, M.J. Project Score database: A resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dede, M.; McLaughlin, M.; Kim, E.; Hart, T. Multiplex enCas12a screens detect functional buffering among paralogs otherwise masked in monogenic Cas9 knockout screens. Genome Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.P.; Tan, J.; Gascoigne, K.E.; Haverty, P.M.; Forrest, W.F.; Costa, M.R.; Martin, S.E. Multiple-gene targeting and mismatch tolerance can confound analysis of genome-wide pooled CRISPR screens. Genome Biol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobzhansky, T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946, 31, 269. [Google Scholar]
- Haince, J.F.; Rouleau, M.; Hendzel, M.J.; Masson, J.Y.; Poirier, G.G. Targeting poly(ADP-ribosyl)ation: A promising approach in cancer therapy. Trends Mol. Med. 2005, 11, 456–463. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009. [Google Scholar] [CrossRef] [Green Version]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Sigorski, D.; Iżycka-Świeszewska, E.; Bodnar, L. Poly(ADP-Ribose) Polymerase Inhibitors in Prostate Cancer: Molecular Mechanisms, and Preclinical and Clinical Data. Target. Oncol. 2020, 15, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, X.; Li, X.; Zhou, Y.; Chen, K. Deep exploration of PARP inhibitors in breast cancer: Monotherapy and combination therapy. J. Int. Med. Res. 2021, 49. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
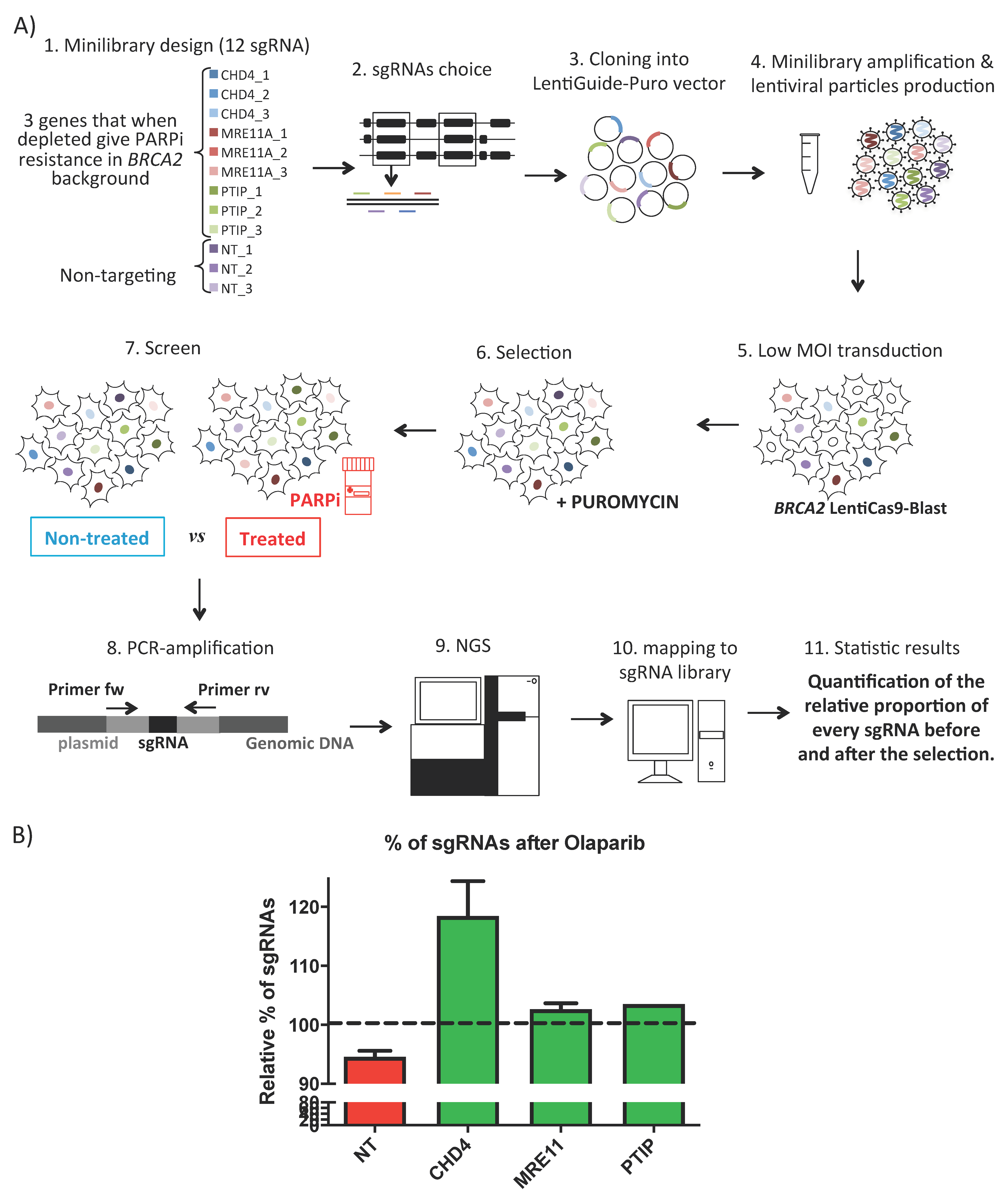
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaite, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018. [Google Scholar] [CrossRef]
- Hewitt, G.; Borel, V.; Segura-Bayona, S.; Takaki, T.; Ruis, P.; Bellelli, R.; Lehmann, L.C.; Sommerova, L.; Vancevska, A.; Tomas-Loba, A.; et al. Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [Green Version]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Nogueira, M.F.; Buss, C.G.; Krill-Burger, J.M.; Wawer, M.J.; Malolepsza, E.; Berger, A.C.; Choi, P.S.; Shih, J.; Taylor, A.M.; et al. Genome-scale analysis identifies paralog lethality as a vulnerability of chromosome 1p loss in cancer. Nat. Genet. 2018. [Google Scholar] [CrossRef]
- Neggers, J.E.; Paolella, B.R.; Asfaw, A.; Rothberg, M.V.; Skipper, T.A.; Yang, A.; Kalekar, R.L.; Krill-Burger, J.M.; Dharia, N.V.; Kugener, G.; et al. Synthetic Lethal Interaction between the ESCRT Paralog Enzymes VPS4A and VPS4B in Cancers Harboring Loss of Chromosome 18q or 16q. Cell Rep. 2020. [Google Scholar] [CrossRef]
- Van der Lelij, P.; Newman, J.A.; Lieb, S.; Jude, J.; Katis, V.; Hoffmann, T.; Hinterndorfer, M.; Bader, G.; Kraut, N.; Pearson, M.A.; et al. STAG1 vulnerabilities for exploiting cohesin synthetic lethality in STAG2-deficient cancers. Life Sci. Alliance 2020. [Google Scholar] [CrossRef]
- Jia, J.; Zhu, F.; Ma, X.; Cao, Z.W.; Li, Y.X.; Chen, Y.Z. Mechanisms of drug combinations: Interaction and network perspectives. Nat. Rev. Drug Discov. 2009. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Wu, H.J.; Ge, J.Y.; Zeid, R.; Harris, I.S.; Jovanović, B.; Murphy, K.; Wang, B.; Qiu, X.; Endress, J.E.; et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.F.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020. [Google Scholar] [CrossRef]
- Hou, P.; Wu, C.; Wang, Y.; Qi, R.; Bhavanasi, D.; Zuo, Z.; Dos Santos, C.; Chen, S.; Chen, Y.; Zheng, H.; et al. A genome-wide CRISPR screen identifies genes critical for resistance to FLT3 inhibitor AC220. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daggubati, V.; Hochstetler, J.; Bommireddy, A.; Choudhury, A.; Krup, A.L.; Choksi, P.K.; Tong, P.; Li, A.; Xu, L.; Reiter, J.F.; et al. Smoothened-activating lipids drive resistance to CDK4/6 inhibition in Hedgehog-associated medulloblastoma cells and preclinical models. J. Clin. Investig. 2021. [Google Scholar] [CrossRef]
- Hustedt, N.; Álvarez-Quilón, A.; McEwan, A.; Yuan, J.Y.; Cho, T.; Koob, L.; Hart, T.; Durocher, D. A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sánchez-Rivera, F.J.; Westcott, P.M.K.; Mercer, K.L.; Bhutkar, A.; Muir, A.; González Robles, T.J.; Lamboy Rodríguez, S.; Liao, L.Z.; Ng, S.R.; et al. Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nat. Cancer 2020. [Google Scholar] [CrossRef]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 2018. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, B.A.; Bennett, N.K.; Darch, M.A.; Yu, K.; Nguyen, M.K.; Pucciarelli, D.; Nelson, M.; Horlbeck, M.A.; Gilbert, L.A.; Hyun, W.; et al. A high-throughput screen of real-time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016. [Google Scholar] [CrossRef] [Green Version]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Pierce, S.E.; Li, A.; Spees, K.; Anderson, G.R.; Seoane, J.A.; Lo, Y.H.; Dubreuil, M.; Olivas, M.; Kamber, R.A.; et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 2020. [Google Scholar] [CrossRef]
- Takahashi, N.; Cho, P.; Selfors, L.M.; Kuiken, H.J.; Kaul, R.; Fujiwara, T.; Harris, I.S.; Zhang, T.; Gygi, S.P.; Brugge, J.S. 3D Culture Models with CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol. Cell 2020. [Google Scholar] [CrossRef]
- Xian, L.; Chia, L.; Georgess, D.; Luo, L.; Shuai, S.; Ewald, A.J.; Resar, L.M.S. Genetic engineering of primary mouse intestinal organoids using magnetic nanoparticle transduction viral vectors for frozen sectioning. J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [Green Version]
- Kashfi, H.; Jinks, N.; Nateri, A.S. Generating and utilizing murine cas9-expressing intestinal organoids for large-scale knockout genetic screening. In Methods in Molecular Biology; Humana: New York, NY, USA, 2020. [Google Scholar]
- Ringel, T.; Frey, N.; Ringnalda, F.; Janjuha, S.; Cherkaoui, S.; Butz, S.; Srivatsa, S.; Pirkl, M.; Russo, G.; Villiger, L.; et al. Genome-Scale CRISPR Screening in Human Intestinal Organoids Identifies Drivers of TGF-β Resistance. Cell Stem Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Michels, B.E.; Mosa, M.H.; Streibl, B.I.; Zhan, T.; Menche, C.; Abou-El-Ardat, K.; Darvishi, T.; Członka, E.; Wagner, S.; Winter, J.; et al. Pooled In Vitro and In Vivo CRISPR-Cas9 Screening Identifies Tumor Suppressors in Human Colon Organoids. Cell Stem Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Cortez, J.T.; Montauti, E.; Shifrut, E.; Gatchalian, J.; Zhang, Y.; Shaked, O.; Xu, Y.; Roth, T.L.; Simeonov, D.R.; Zhang, Y.; et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Huang, Q.; Luster, T.A.; Hu, H.; Zhang, H.; Ng, W.L.; Khodadadi-Jamayran, A.; Wang, W.; Chen, T.; Deng, J.; et al. In vivo epigenetic crispr screen identifies asf1a as an immunotherapeutic target in kras-mutant lung adenocarcinoma. Cancer Discov. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. J. Clean. Prod. 2020. [Google Scholar] [CrossRef]
- Szlachta, K.; Kuscu, C.; Tufan, T.; Adair, S.J.; Shang, S.; Michaels, A.D.; Mullen, M.G.; Fischer, N.L.; Yang, J.; Liu, L.; et al. CRISPR knockout screening identifies combinatorial drug targets in pancreatic cancer and models cellular drug response. Nat. Commun. 2018. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Rushworth, L.K.; Harle, V.; Repiscak, P.; Clark, W.; Shaw, R.; Hall, H.; Bushell, M.; Leung, H.Y.; Patel, R. In vivo CRISPR/Cas9 knockout screen: TCEAL1 silencing enhances docetaxel efficacy in prostate cancer. Life Sci. Alliance 2020. [Google Scholar] [CrossRef]
- Braun, C.J.; Bruno, P.M.; Horlbeck, M.A.; Gilbert, L.A.; Weissman, J.S.; Hemann, M.T. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.; Yao, S.; Zhang, Y.; Ye, Y.; Li, C.; Hu, L.; Sun, Y.; Huang, H.Y.; Ji, H. In vivo miRNA knockout screening identifies miR-190b as a novel tumor suppressor. PLoS Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Fröse, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Okabe, A.; Kaneda, A. Transcriptional dysregulation by aberrant enhancer activation and rewiring in cancer. Cancer Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015. [Google Scholar] [CrossRef] [Green Version]
- Diao, Y.; Li, B.; Meng, Z.; Jung, I.; Lee, A.Y.; Dixon, J.; Maliskova, L.; Guan, K.L.; Shen, Y.; Ren, B. A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Fulco, C.P.; Munschauer, M.; Anyoha, R.; Munson, G.; Grossman, S.R.; Perez, E.M.; Kane, M.; Cleary, B.; Lander, E.S.; Engreitz, J.M. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 2016. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Rajagopal, N.; Srinivasan, S.; Kooshesh, K.; Guo, Y.; Edwards, M.D.; Banerjee, B.; Syed, T.; Emons, B.J.M.; Gifford, D.K.; Sherwood, R.I. High-throughput mapping of regulatory DNA. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Wright, J.; Zheng, K.; Shalem, O.; Fontanillas, P.; Joung, J.; Cheng, C.; Regev, A.; Zhang, F. High-resolution interrogation of functional elements in the noncoding genome. Science 2016. [Google Scholar] [CrossRef] [Green Version]
- Diao, Y.; Fang, R.; Li, B.; Meng, Z.; Yu, J.; Qiu, Y.; Lin, K.C.; Huang, H.; Liu, T.; Marina, R.J.; et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 2017. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, M.; Findlay, G.M.; McKenna, A.; Milbank, J.H.; Lee, C.; Zhang, M.D.; Cusanovich, D.A.; Shendure, J. CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017. [Google Scholar] [CrossRef]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S.; et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Yang, L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef]
- John Liu, S.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020. [Google Scholar] [CrossRef]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Z.; Wang, Y.; Guo, Y.; Xu, P.; Yuan, P.; Liu, Z.; He, Y.; Wei, W. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Nana-Sinkam, S.P.; Croce, C.M. MicroRNA regulation of tumorigenesis, cancer progression and interpatient heterogeneity: Towards clinical use. Genome Biol. 2014, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.; Hu, R.; Mosbruger, T.L.; Dahlem, T.J.; Stephens, W.Z.; Rao, D.S.; Round, J.L.; O’Connell, R.M. Genome-wide CRISPR-Cas9 screen identifies microRNAs that regulate myeloid leukemia cell growth. PLoS ONE 2016. [Google Scholar] [CrossRef]
- Kurata, J.S.; Lin, R.J. MicroRNA-focused CRISPR-Cas9 library screen reveals fitness-associated miRNAs. RNA 2018. [Google Scholar] [CrossRef] [Green Version]
- Potts, M.A.; McDonald, J.A.; Sutherland, K.D.; Herold, M.J. Critical cancer vulnerabilities identified by unbiased CRISPR/Cas9 screens inform on efficient cancer Immunotherapy. Eur. J. Immunol. 2020, 50, 1871–1884. [Google Scholar] [CrossRef]
- Liu, D.; Zhao, X.; Tang, A.; Xu, X.; Liu, S.; Zha, L.; Ma, W.; Zheng, J.; Shi, M. CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188378. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroblewska, A.; Dhainaut, M.; Ben-Zvi, B.; Rose, S.A.; Park, E.S.; Amir, E.A.D.; Bektesevic, A.; Baccarini, A.; Merad, M.; Rahman, A.H.; et al. Protein Barcodes Enable High-Dimensional Single-Cell CRISPR Screens. Cell 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, D.; Singh, A.; Schmid-Burgk, J.L.; Carlson, R.J.; Mezger, A.; Garrity, A.J.; Zhang, F.; Blainey, P.C. Optical Pooled Screens in Human Cells. Cell 2019. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| System | Library | Model | Analysis | SLI between |
|---|---|---|---|---|
| CRISPR-based double KO [97] | 21,321 pairs of drug targets | K562 leukemia cells | casTLE | DNA repair proteins APEX1 and ATM, anti-apoptotic BCL2L1 and MCL1 |
| CRISPR-based double KO [62] | 119 KRAS interactor targets | lung adenocarcinoma cells | UPGMA scipy statsmodels | RAS adhesion controller RADIL and endocytosis regulator RIN1, RAP1GDS1 and RHOA |
| Enhance alisertib Aurora-A inhibitor activity [69] | 507 kinase targets | Breast cancer cells | MAGeCK | GSG2 inhibition (interfering with AURORA-B) significantly decreased tumor growth in vitro and in vivo |
| Aim | Library | Model | Treatment | Analysis | Targets |
|---|---|---|---|---|---|
| Resistances to FLT3 inhibitor [102] | GeCKO | MV4-11 acute myeloid leukemia cells | Quizartinib | Self-calculated | Expression of SPRY3 and GSK3A was significantly decreased in resistant cells |
| Resistances to multi-targeted tyrosine kinase inhibitors (TKIs) [70] | Customized library (18,000 targets) | clear cell renal cell carcinoma (ccRCC) | Sunitinib | Self-calculated | farnesyltransferase expression as a factor of sunitinib resistance |
| Asparaginase responses [63] | GeCKO | acute leukemia cells (ALC) | Asparaginase | MAGeCK v0.5.7 | Wnt signalling induced asparaginase sensitivity in resistant ALC |
| Synergizes with metformin [66] | Brunello | U251 cells | Metformin | MAGeCK | Metformin and CDK4/6 inhibitor combination as tumoral therapy |
| Abemaciclib resistance CRISPR and CRISPRi screen [103] | Brunello and CRISPRi-v2 | Hedgehog associated medulloblastoma cells | Abemaciclib | MAGeCK -VSIPR | Hedgehog signaling in neuroblastoma depends on smoothened-activating sterol lipids |
| Molecular pathways depending on ataxia-telangiectasia and Rad3-related (ATR) kinase [104] | TKOv1 and TKOv3 | colon carcinoma HCT116, HeLa and a p53-mutated clone of RPE1 hTERT cells | ATR inhibitors VE-821 and AZD6738 | MAGeCK and drugZ | DNA replication, DNA repair and cell cycle regulators give hypersensitive to ATR inhibitors. POLE3/POLE4 proteins are potential biomarkers for ATR processes. |
| Druggable targets in RNF43-mutant pancreatic adenocarcinomas [67] | TKO gRNA library | HPAF-II human pancreatic ductal adenocarcinoma cell line | - | BAGEL algorithm | Wnt receptor Frizzled-5 (FZD5) |
| Druggable targets in Keap1a-mutant or NRF2-hyperactive tumors [105] | 4,915 druggable targets library | Murine lung adenocarcinoma (LUAD) KrasG12D/+; p53−/− (KP) versus KrasG12D/+; p53−/−; Keap1−/− (KPK) cell lines | - | RSEM, JADE algorithm and GSEA | SLC33A1 and unfolded protein response related genes are novel targets for patients harboring KEAP1-mutant or NRF2-hyperactivated tumors |
| Aim | Library | Model | Method | Analysis | Targets |
|---|---|---|---|---|---|
| Cancer biomarker and therapy [110] | Customized CRISPR ko library | H23 LUAD 2D cell line and 3D spheroids |
3D versus 2D cultures | computed t-value scores |
p53 and Ras are 3D hits, IGF1R expression/dependency and KRAS mutation may serve as biomarkers |
| NRF2 hyperactivation-induced spheroid growth and NRF2-hyperactivated tumors [111] | Customized 1,500 NRF2-hyperactivated related gene targets library | A549 and H1437 LUAD 2D cell line and 3D spheroids |
3D versus 2D cultures | MAGeCK-VISPR | In spheroids, loss of TSC1 enhances inner clearance and depletion of GPX4 enhances proliferation |
| Resistances to TGF-β-mediated growth restriction [114] | 283 potential tumor suppressor genes customized library and the Brunello library | Human small intestinal (hSI) organoids | wild-type versus APC mutant and APC and TP53 double mutant human intestinal organoids | MAGeCK V0.5.4 | Multiple subunits of the tumor-suppressive SWI/SNF chromatin remodeling complex |
| Tumor drivers in colorectal cancer (CRC) [115] | 85 tumor suppressor genes customized library | Pre-malignant organoids with APC−/−; KRASG12D mutations | Primary organoids versus cancer cell lines |
CRISPR- ERA | TGFBR2 and CRC growth mediators |
| Aim | Library | Model | Method | Analysis | Targets |
|---|---|---|---|---|---|
| Loss-of-function screen in tumor growth and metastasis [117] | mGeCKOa | Tumor-inducible non-small-cell lung cancer (NSCLC) cell line | Metastasis versus primary tumors | GSEA | Mutations that inactivate apoptosis and Nf2, Trim72, Ptges2 genes in primary tumor cells, and Ube2g2 mutations in metastasis |
| Epigenetic regulators of tumor immunity [118] | Customized epigenetic sgRNA subpooled | Murine KrasG12D/Trp53−/− LUAD | Anti-PD-1 or isotype control treatments | DESeq2, MAGeCK and GSEA | Asf1a reduced in tumors of WT mice treated with anti–PD-1 |
| Cellular composition and architecture of cutaneous squamous cell carcinoma (cSCC) [119] | Low expressed xenograft genes subpooled | Several cSCC cell lines | Tumor analysis | STARS | TSK-enriched integrin signaling genes ITGB1, FERMT1 and CD151. |
| Non-small cell lung cancers treatment [68] | Customized epigenetic sgRNA subpooled | KP, non-small cell lung cancers cells | Tumor analysis | GSEA, MSigDB and DESeq2 | Npm1 |
| Synergistic lethal drug interactions with MEK signalling pathway inhibitors to treat pancreatic ductal adenocarcinoma (PDAC) [120] | Nuclear subpooled [15] | PDX366 cells from pancreatic patients | Trametinib treatment | DREBIC | CENPE and RRM1 inhibition are sensitizers to trametinib |
| SL combinatorial target with gemcitabine to treat PDAC [121] | Customized epigenetic sgRNA subpooled | PDX366 cells from pancreatic patients | Gemcitabine treatment | MAGeCK v0.5.2. | Inhibition of PRMT5 increases cytotoxicity to gemcitabine |
| Find mechanisms of resistance to docetaxel to treat metastatic prostate cancer [122] | GeCKOv2A | deficient Pten and Spry2 model cells | Docetaxel treatment | MAGeCK v0.5.6. | Suppression of TCEAL1 enhances tumor sensitivity to docetaxel |
| Gene activation screen in vivo [123] | CRISPRa sgRNA targeting 25 DNA damage regulators | dCas9-VP64-expressing Bcr-Abl– driven murine acute B-cell lymphoblastic leukemia cells | Temozolomide treatment | DESeq and self- calculated | Transcriptional activation of tumor suppressor Chek2 sensitizes tumor cells to temozolomide |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castells-Roca, L.; Tejero, E.; Rodríguez-Santiago, B.; Surrallés, J. CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer. Cancers 2021, 13, 1591. https://doi.org/10.3390/cancers13071591
Castells-Roca L, Tejero E, Rodríguez-Santiago B, Surrallés J. CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer. Cancers. 2021; 13(7):1591. https://doi.org/10.3390/cancers13071591
Chicago/Turabian StyleCastells-Roca, Laia, Eudald Tejero, Benjamín Rodríguez-Santiago, and Jordi Surrallés. 2021. "CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer" Cancers 13, no. 7: 1591. https://doi.org/10.3390/cancers13071591
APA StyleCastells-Roca, L., Tejero, E., Rodríguez-Santiago, B., & Surrallés, J. (2021). CRISPR Screens in Synthetic Lethality and Combinatorial Therapies for Cancer. Cancers, 13(7), 1591. https://doi.org/10.3390/cancers13071591