
Long, Noncoding RNA Dysregulation in Glioblastoma

 , ,
, , 
Abstract
Simple Summary
Abstract
1. Noncoding RNAs Are Transcribed from a Large Proportion of the Genome
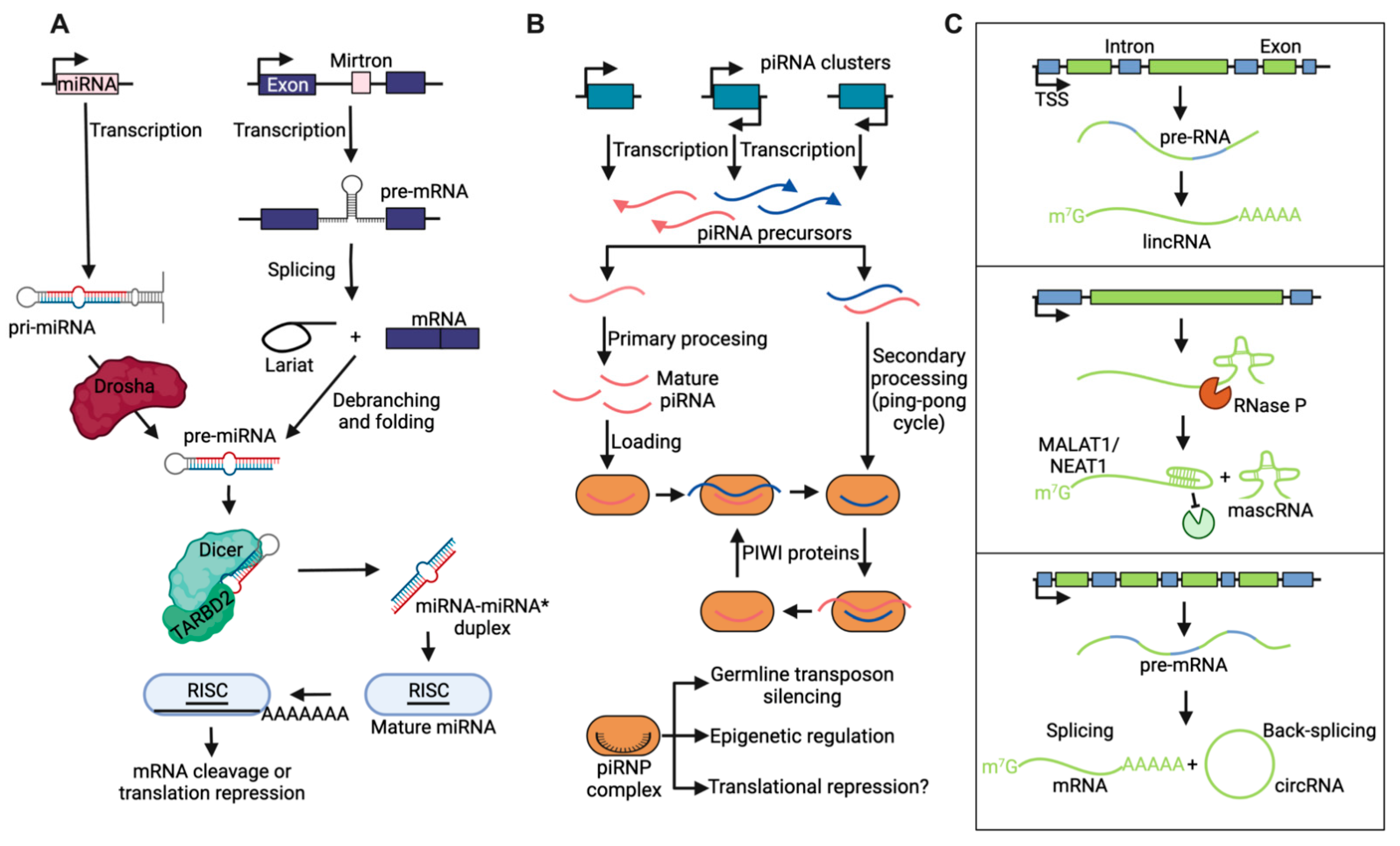
1.1. Noncoding RNA Nomenclature and Biogenesis
1.2. Evolutionary Conservation of Noncoding Transcripts
1.3. Long, Noncoding RNAs in the Central Nervous System
2. General Mechanisms of lncRNAs
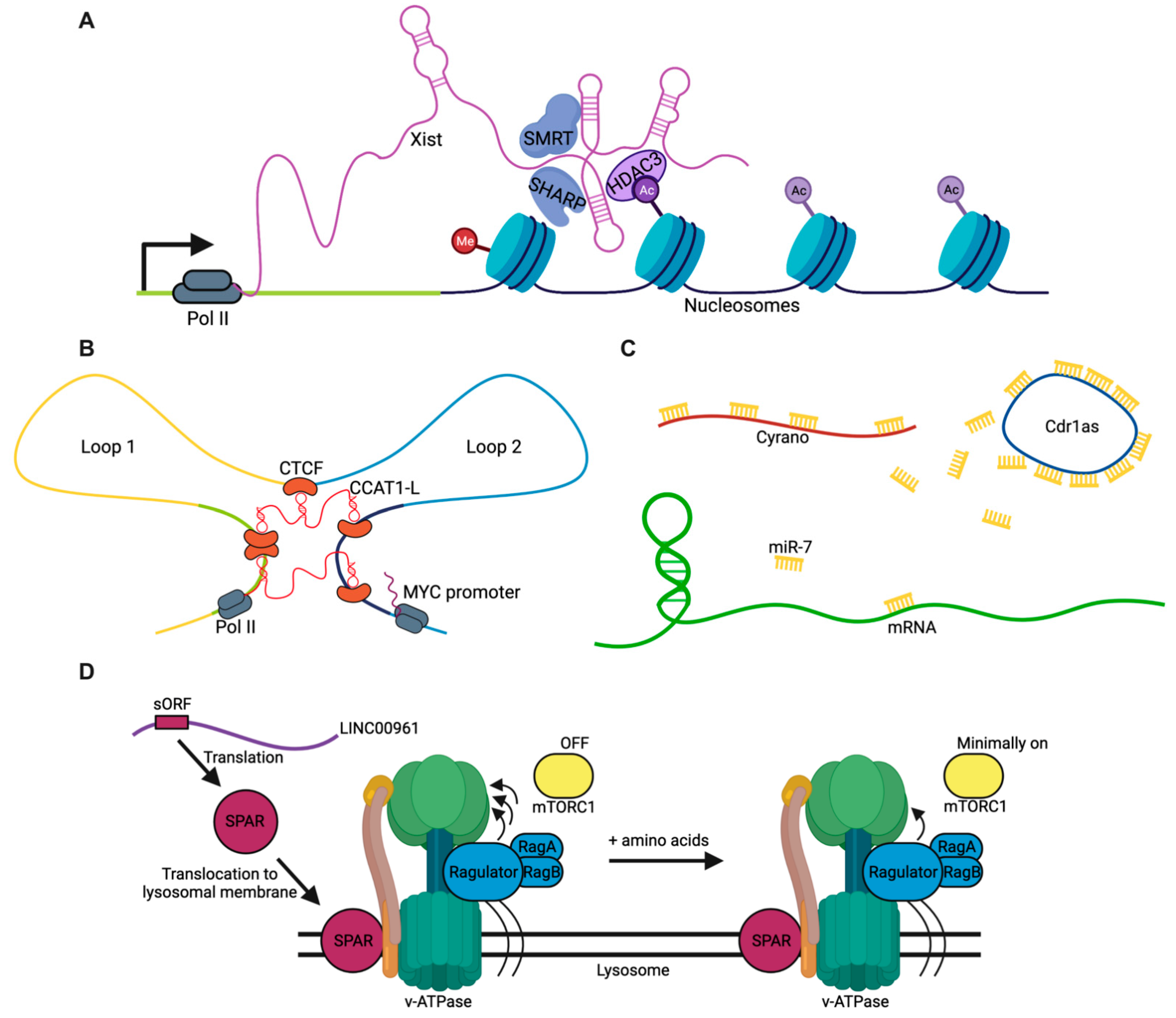
2.1. Guides
2.2. Scaffolds
2.3. Sponges
2.4. Peptides
2.5. Considering Structural Determinants of lncRNA Function
3. Glioblastoma Is an Aggressive, Fatal Brain Tumor
4. Tumor Heterogeneity and Therapeutic Resistance in GBM
4.1. Defining Transcriptional Subtypes in GBM
4.2. Neurodevelopmental Signatures in GBM Heterogeneity and Therapeutic Resistance
4.3. Influence of Tumor Microenvironment
5. Noncoding Aberrations Are Understudied Molecular Players in GBM
5.1. Somatic Drivers and Structural Variants in lncRNAs
5.2. lncRNA Expression Deregulation
6. RNA-Based Therapeutics
7. Conclusions and Perspectives
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef]
- Khanduja, J.S.; Calvo, I.A.; Joh, R.I.; Hill, I.T.; Motamedi, M. Nuclear Noncoding RNAs and Genome Stability. Mol. Cell 2016, 63, 7–20. [Google Scholar] [CrossRef]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010, 11, 559–571. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for argonautes. Nat. Rev. Genet. 2011, 12, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Aravin, A.A.; Sachidanandam, R.; Girard, A.; Fejes-Toth, K.; Hannon, G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007, 316, 744–747. [Google Scholar] [CrossRef]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete Small RNA-Generating Loci as Master Regulators of Transposon Activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Vaughn, I.W.; McManus, M.T. Pervasive Transcription of the Human Genome Produces Thousands of Previously Unidentified Long Intergenic Noncoding RNAs. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Othoum, G.; Coonrod, E.; Zhao, S.; Dang, H.X.; Maher, C.A. Pan-cancer proteogenomic analysis reveals long and circular noncoding RNAs encoding peptides. NAR Cancer 2020, 2, 1–11. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, K.; Xu, X.; Yang, Y.; Yan, S.; Wei, P.; Liu, H.; Xu, J.; Xiao, F.; Zhou, H.; et al. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y.; Chang, H.Y. Long Noncoding RNAs: Molecular Modalities to Organismal Functions. Annu. Rev. Biochem. 2020, 89, 283–308. [Google Scholar] [CrossRef]
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigó, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef]
- Li, X.; Fu, X.D. Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat. Rev. Genet. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2019. [Google Scholar] [CrossRef]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Chen, J.Y.; Liang, Z.; Luo, D.; Chen, G.; Lu, Z.J.; Chen, Y.; Zhou, B.; Li, H.; Du, X.; et al. Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 2019, 178, 107–121.e18. [Google Scholar] [CrossRef]
- Holmes, Z.E.; Hamilton, D.J.; Hwang, T.; Parsonnet, N.V.; Rinn, J.L.; Wuttke, D.S.; Batey, R.T. The Sox2 transcription factor binds RNA. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef]
- Kirk, J.M.; Kim, S.O.; Inoue, K.; Smola, M.J.; Lee, D.M.; Schertzer, M.D.; Wooten, J.S.; Baker, A.R.; Sprague, D.; Collins, D.W.; et al. Functional classification of long non-coding RNAs by k-mer content. Nat. Genet. 2018, 50, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.; Caudron-Herger, M.; Diederichs, S. RNA motifs and combinatorial prediction of interactions, stability and localization of noncoding RNAs. Nat. Struct. Mol. Biol. 2018, 25, 1070–1076. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Koonin, E.V. Functional Long Non-coding RNAs Evolve from Junk Transcripts. Cell 2020, 183, 1151–1161. [Google Scholar] [CrossRef]
- Stoltzfus, A. Constructive neutral evolution: Exploring evolutionary theory’s curious disconnect. Biol. Direct 2012, 7, 1–13. [Google Scholar] [CrossRef]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wingo, A.P.; Dammer, E.B.; Breen, M.S.; Logsdon, B.A.; Duong, D.M.; Troncosco, J.C.; Thambisetty, M.; Beach, T.G.; Serrano, G.E.; Reiman, E.M.; et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Belgard, T.G.; Marques, A.C.; Oliver, P.L.; Abaan, H.O.; Sirey, T.M.; Hoerder-Suabedissen, A.; García-Moreno, F.; Molnár, Z.; Margulies, E.H.; Ponting, C.P. A transcriptomic atlas of mouse neocortical layers. Neuron 2011, 71, 605–616. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef]
- Molyneaux, B.J.; Goff, L.A.; Brettler, A.C.; Chen, H.H.; Brown, J.R.; Hrvatin, S.; Rinn, J.L.; Arlotta, P. DeCoN: Genome-wide analysis of invivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron 2015, 85, 275–288. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.D.; Diaz, A.; Nellore, A.; Delgado, R.N.; Park, K.Y.; Gonzales-Roybal, G.; Oldham, M.C.; Song, J.S.; Lim, D.A. Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell 2013, 12, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Li, W.; Tian, H.; Hu, T.; Wang, L.; Lin, Y.; Li, Y.; Huang, H.; Sun, F. Sequential expression of long noncoding RNA as mRNA gene expression in specific stages of mouse spermatogenesis. Sci. Rep. 2014, 4, 1–6. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, T.; Zhou, W.; Li, J.; Li, X.; Wang, Q.; Jin, X.; Yin, J.; Chen, L.; Zhang, Y.; et al. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Agirre, X.; Meydan, C.; Jiang, Y.; Garate, L.; Doane, A.S.; Li, Z.; Verma, A.; Paiva, B.; Martín-Subero, J.I.; Elemento, O.; et al. Long non-coding RNAs discriminate the stages and gene regulatory states of human humoral immune response. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Xu, C.L.; Sang, B.; Liu, G.Z.; Li, J.M.; Zhang, X.D.; Liu, L.X.; Thorne, R.F.; Wu, M. SENEBLOC, a long non-coding RNA suppresses senescence via p53-dependent and independent mechanisms. Nucleic Acids Res. 2020, 48, 3089–3102. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef]
- Chu, C.; Zhang, Q.C.; Da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef]
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 2018, 174, 350–362.e17. [Google Scholar] [CrossRef]
- Matsumoto, A.; Pasut, A.; Matsumoto, M.; Yamashita, R.; Fung, J.; Monteleone, E.; Saghatelian, A.; Nakayama, K.I.; Clohessy, J.G.; Pandolfi, P.P. MTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 2017, 541, 228–232. [Google Scholar] [CrossRef]
- Lin, A.; Hu, Q.; Li, C.; Xing, Z.; Ma, G.; Wang, C.; Li, J.; Ye, Y.; Yao, J.; Liang, K.; et al. The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 2017, 19, 238–251. [Google Scholar] [CrossRef]
- Schertzer, M.D.; Braceros, K.C.A.; Starmer, J.; Cherney, R.E.; Lee, D.M.; Salazar, G.; Justice, M.; Bischoff, S.R.; Cowley, D.O.; Ariel, P.; et al. lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol. Cell 2019, 75, 523–537.e10. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Hwang, T.; Gooding, A.R.; Goodrich, K.J.; Rinn, J.L.; Cech, T.R. RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat. Genet. 2020, 52, 931–938. [Google Scholar] [CrossRef]
- Grossi, E.; Raimondi, I.; Goñi, E.; González, J.; Marchese, F.P.; Chapaprieta, V.; Martín-Subero, J.I.; Guo, S.; Huarte, M. A lncRNA-SWI/SNF complex crosstalk controls transcriptional activation at specific promoter regions. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Souquere, S.; Chujo, T.; Kobelke, S.; Chong, Y.S.; Fox, A.H.; Bond, C.S.; Nakagawa, S.; Pierron, G.; Hirose, T. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 2018, 70, 1038–1053.e7. [Google Scholar] [CrossRef]
- Uroda, T.; Anastasakou, E.; Rossi, A.; Teulon, J.M.; Pellequer, J.L.; Annibale, P.; Pessey, O.; Inga, A.; Chillón, I.; Marcia, M. Conserved Pseudoknots in lncRNA MEG3 Are Essential for Stimulation of the p53 Pathway. Mol. Cell 2019, 75, 982–995.e9. [Google Scholar] [CrossRef]
- Munschauer, M.; Nguyen, C.T.; Sirokman, K.; Hartigan, C.R.; Hogstrom, L.; Engreitz, J.M.; Ulirsch, J.C.; Fulco, C.P.; Subramanian, V.; Chen, J.; et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 2018, 561, 132–136. [Google Scholar] [CrossRef]
- Lee, S.; Kopp, F.; Chang, T.C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.-j.; Ju, H.-q.; Liu, G.-p.; Tian, T.; Ma, G.-l.; Lu, Y.-x.; Liu, Z.-x.; Pan, R.-l.; Li, R.-h.; Piao, H.-l.; et al. LncRNA CamK-A Regulates Ca2+-Signaling-Mediated Tumor Microenvironment Remodeling. Mol. Cell 2018, 72, 71–83.e7. [Google Scholar] [CrossRef]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef]
- Ganser, L.R.; Kelly, M.L.; Herschlag, D.; Al-Hashimi, H.M. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 2019, 20, 25–27. [Google Scholar] [CrossRef]
- Cammas, A.; Millevoi, S. RNA G-quadruplexes: Emerging mechanisms in disease. Nucleic Acids Res. 2017, 45, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Herviou, P.; Le Bras, M.; Dumas, L.; Hieblot, C.; Gilhodes, J.; Cioci, G.; Hugnot, J.P.; Ameadan, A.; Guillonneau, F.; Dassi, E.; et al. hnRNP H/F drive RNA G-quadruplex-mediated translation linked to genomic instability and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, M.; Ma, Y.; Nagasawa, K.; Toyoshima, F. A G-quadruplex structure at the 5′ end of the H19 coding region regulates H19 transcription. Sci. Rep. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro. Oncol. 2019, 21, V1–V100. [Google Scholar] [CrossRef]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef] [PubMed]
- Körber, V.; Yang, J.; Barah, P.; Wu, Y.; Stichel, D.; Gu, Z.; Fletcher, M.N.C.; Jones, D.; Hentschel, B.; Lamszus, K.; et al. Evolutionary Trajectories of IDHWT Glioblastomas Reveal a Common Path of Early Tumorigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell 2019, 1–13. [Google Scholar] [CrossRef]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Komotar, R.J.; Otten, M.L.; Moise, G.; Connolly, E.S. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma—A critical review. Clin. Med. Oncol. 2008, 2, 421–422. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 1–15. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef]
- Wang, L.; Babikir, H.; Müller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e6. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D.; et al. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Paturu, M.R.; Patel, B.; Cain, M.D.; Mahlokozera, T.; Yang, A.B.; Lin, T.-H.; Leuthardt, E.C.; Yano, H.; Song, S.-K.; et al. Therapeutic enhancement of blood–brain and blood–tumor barriers permeability by laser interstitial thermal therapy. Neuro-Oncol. Adv. 2020, 2, 1–12. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
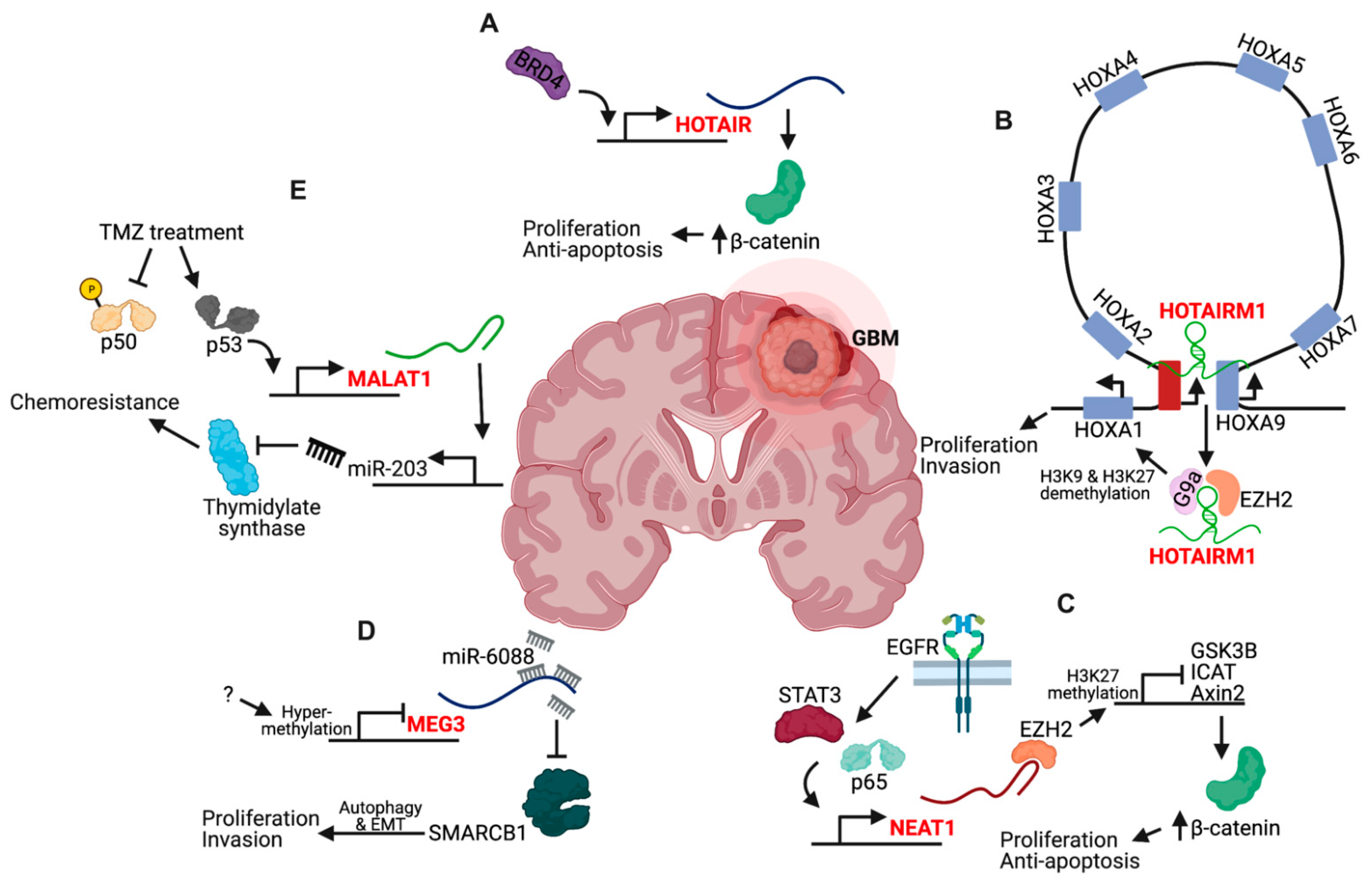
- Pastori, C.; Kapranov, P.; Penas, C.; Peschansky, V.; Volmar, C.H.; Sarkaria, J.N.; Bregy, A.; Komotar, R.; Laurent, G.S.; Ayad, N.G.; et al. The bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl. Acad. Sci. USA 2015, 112, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ren, Y.; Zhang, J.; Zhang, C.; Zhang, K.; Han, L.; Kong, L.; Wei, J.; Chen, L.; Yang, J.; et al. HOTAIR is a therapeutic target in glioblastoma. Oncotarget 2015, 6, 8353–8365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lian, Z.; Padden, C.; Gerstein, M.B.; Rozowsky, J.; Snyder, M.; Gingeras, T.R.; Kapranov, P.; Weissman, S.M.; Newburger, P.E. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood 2009, 113, 2526–2534. [Google Scholar] [CrossRef]
- Stackhouse, C.T.; Gillespie, G.Y.; Willey, C.D. Exploring the Roles of lncRNAs in GBM Pathophysiology and Their Therapeutic Potential. Cells 2020, 9, 2369. [Google Scholar] [CrossRef]
- Xie, P.; Li, X.; Chen, R.; Liu, Y.; Liu, D.C.; Liu, W.; Cui, G.; Xu, J. Upregulation of HOTAIRM1 increases migration and invasion by glioblastoma cells. Aging 2021, 13, 2348–2364. [Google Scholar] [CrossRef]
- Li, Q.; Dong, C.; Cui, J.; Wang, Y.; Hong, X. Over-expressed lncRNA HOTAIRM1 promotes tumor growth and invasion through up-regulating HOXA1 and sequestering G9a/EZH2/Dnmts away from the HOXA1 gene in glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2018, 37, 1–15. [Google Scholar] [CrossRef]
- Shi, T.; Guo, D.; Xu, H.; Su, G.; Chen, J.; Zhao, Z.; Shi, J.; Wedemeyer, M.; Attenello, F.; Zhang, L.; et al. HOTAIRM1, an enhancer lncRNA, promotes glioma proliferation by regulating long-range chromatin interactions within HOXA cluster genes. Mol. Biol. Rep. 2020, 47, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Cai, J.; Wang, Q.; Wang, Y.; Liu, M.; Yang, J.; Zhou, J.; Kang, C.; Li, M.; Jiang, C. Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/b-catenin pathway by scaffolding EZH2. Clin. Cancer Res. 2018, 24, 684–695. [Google Scholar] [CrossRef]
- Lulli, V.; Buccarelli, M.; Ilari, R.; Castellani, G.; De Dominicis, C.; Di Giamberardino, A.; D′Alessandris, Q.G.; Giannetti, S.; Martini, M.; Stumpo, V.; et al. Mir-370-3p impairs glioblastoma stem-like cell malignancy regulating a complex interplay between HMGA2/HIF1A and the oncogenic long non-coding RNA (LncRNA) neat1. Int. J. Mol. Sci. 2020, 21, 3610. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Zang, J.; Zheng, M.-H.; Zhang, Y.-F.; Yue, K.-Y.; Cao, X.-L.; Cao, Y.; Li, X.-X.; Han, H.; Jiang, X.-F.; et al. Temozolomide Treatment Induces HMGB1 to Promote the Formation of Glioma Stem Cells via the TLR2/NEAT1/Wnt Pathway in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Zhang, X.; Rice, K.; Wang, Y.; Chen, W.; Zhong, Y.; Nakayama, Y.; Zhou, Y.; Klibanski, A. Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: Isoform structure, expression, and functions. Endocrinology 2010, 151, 939–947. [Google Scholar] [CrossRef]
- Gong, X.; Huang, M.Y. Tumor-Suppressive Function of lncRNA-MEG3 in Glioma Cells by Regulating miR-6088/SMARCB1 Axis. Biomed Res. Int. 2020, 2020, 4309161. [Google Scholar] [CrossRef]
- Yang, Z.; Bian, E.; Xu, Y.; Ji, X.; Tang, F.; Ma, C.; Wang, H.; Zhao, B. Meg3 induces EMT and invasion of glioma cells via autophagy. OncoTargets Ther. 2020, 13, 989–1000. [Google Scholar] [CrossRef]
- Buccarelli, M.; Lulli, V.; Giuliani, A.; Signore, M.; Martini, M.; D’Alessandris, Q.G.; Giannetti, S.; Novelli, A.; Ilari, R.; Giurato, G.; et al. Deregulated expression of the imprinted DLK1-DIO3 region in glioblastoma stemlike cells: Tumor suppressor role of lncRNA MEG3. Neuro Oncol. 2020, 22, 1771–1784. [Google Scholar] [CrossRef]
- Liao, K.; Lin, Y.; Gao, W.; Xiao, Z.; Medina, R.; Dmitriev, P.; Cui, J.; Zhuang, Z.; Zhao, X.; Qiu, Y.; et al. Blocking lncRNA MALAT1/miR-199a/ZHX1 Axis Inhibits Glioblastoma Proliferation and Progression. Mol. Ther. Nucleic Acids 2019, 18, 388–399. [Google Scholar] [CrossRef]
- Chen, W.; Xu, X.K.; Li, J.L.; Kong, K.K.; Li, H.; Chen, C.; He, J.; Wang, F.; Li, P.; Ge, X.S.; et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget 2017, 8, 22783–22799. [Google Scholar] [CrossRef]
- Voce, D.J.; Bernal, G.M.; Wu, L.; Crawley, C.D.; Zhang, W.; Mansour, N.M.; Cahill, K.E.; Szymura, S.J.; Uppal, A.; Raleigh, D.R.; et al. Temozolomide treatment induces lncRNA MALAT1 in an NF-κB and p53 codependent manner in Glioblastoma. Cancer Res. 2019, 79, 2536–2548. [Google Scholar] [CrossRef]
- Kim, S.S.; Harford, J.B.; Moghe, M.; Rait, A.; Pirollo, K.F.; Chang, E.H. Targeted nanocomplex carrying siRNA against MALAT1 sensitizes glioblastoma to temozolomide. Nucleic Acids Res. 2018, 46, 1424–1440. [Google Scholar] [CrossRef]
- Peng, Q.; Li, R.; Li, Y.; Xu, X.; Ni, W.; Lin, H.; Ning, L. Prediction of a competing endogenous RNA co-expression network as a prognostic marker in glioblastoma. J. Cell. Mol. Med. 2020, 24, 13346–13355. [Google Scholar] [CrossRef]
- Liu, G.; Liu, D.; Huang, J.; Li, J.; Wang, C.; Liu, G.; Ge, S.; Gong, H. Comprehensive analysis of ceRNA network related to lincRNA in glioblastoma and prediction of clinical prognosis. BMC Cancer 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Liang, R.; Zhi, Y.; Zheng, G.; Zhang, B.; Zhu, H.; Wang, M. Analysis of long non-coding rnas in glioblastoma for prognosis prediction using weighted gene co-expression network analysis, cox regression, and l1-lasso penalization. OncoTargets Ther. 2019, 12, 157–168. [Google Scholar] [CrossRef]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355. [Google Scholar] [CrossRef]
- Liu, S.J.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020, 21, 83. [Google Scholar] [CrossRef]
- Davis, S.; Lollo, B.; Freier, S.; Esau, C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006, 34, 2294–2304. [Google Scholar] [CrossRef]
- Singh, P.; Singh, A.; Shah, S.; Vataliya, J.; Mittal, A.; Chitkara, D. RNA Interference Nanotherapeutics for Treatment of Glioblastoma Multiforme. Mol. Pharm. 2020, 17, 4040–4066. [Google Scholar] [CrossRef] [PubMed]
- Pourgholi, F.; Hajivalili, M.; Farhad, J.N.; Kafil, H.S.; Yousefi, M. Nanoparticles: Novel vehicles in treatment of Glioblastoma. Biomed. Pharmacother. 2016, 77, 98–107. [Google Scholar] [CrossRef]
- Osborn, M.F.; Coles, A.H.; Golebiowski, D.; Echeverria, D.; Moazami, M.P.; Watts, J.K.; Sena-Esteves, M.; Khvorova, A. Efficient gene silencing in brain tumors with hydrophobically modified siRNAs. Mol. Cancer Ther. 2018, 17, 1251–1258. [Google Scholar] [CrossRef]
- Liu, H.M.; Zhang, Y.F.; Xie, Y.D.; Cai, Y.F.; Li, B.Y.; Li, W.; Zeng, L.Y.; Li, Y.L.; Yu, R.T. Hypoxia-responsive ionizable liposome delivery siRNA for glioma therapy. Int. J. Nanomed. 2017, 12, 1065–1083. [Google Scholar] [CrossRef]
- Babae, N.; Bourajjaj, M.; Liu, Y.; Van Beijnum, J.R.; Cerisoli, F.; Scaria, P.V.; Verheul, M.; Van Berkel, M.P.; Pieters, E.H.E.; Van Haastert, R.J.; et al. Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget 2014, 5, 6687–6700. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef]
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I.; et al. Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef]
- Kouri, F.M.; Hurley, L.A.; Daniel, W.L.; Day, E.S.; Hua, Y.; Hao, L.; Peng, C.-Y.; Merkel, T.J.; Queisser, M.A.; Ritner, C.; et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 2015, 29, 732–745. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | lncRNA Signature | mRNA Signature | Functional Ontologies |
|---|---|---|---|
| Endothelia | LINC-MILR1-3, SLC38A3, LINC00152, RP11-401P9.4, MIR4435-HG, LINC00339, AP000459.4, AC127904.2, RP11-161M6.2, RP11-417F21.1, TRIM52-AS1, CTD-2081C10.7, RP11-296I10.3, RP11-532M24.1 | GPR116, ITM2A, C1orf54, GNG11, COL4A1, ECSCR, EMCN, LAMA4, ECM1, RAPGEF4, A2M, IGFBP7, CD93, FLT1, RNF144B | Angiogenesis Regulation of vasculature development Hemostasis Response to oxygen levels Blood coagulation Coagulation Regulation of angiogenesis Response to decreased oxygen levels Response to hypoxia Extracellular matrix organization |
| Radial Glia | Z83001.1, RP11-731J8.2, LINC00943, RP3-418C23.2, RP11-1002K11.1, MAGI2-AS3, RP11-421L21.3, LINC-FZD3-3, LINC-FZD8-1, LINC00263, EIF3J-AS1, LOC646329, LINC-KREMEN1-1, RUSC1-AS1, DGKK | GPX3, ATP1A2, BCAN, MOXD1, LIPG, CLU, FAM107A, ANXA2, VIM, GFAP, PPAP2B, ZFP36L1, GATM, TNC, HES1 | Negative regulation of nervous system development Negative regulation of neuron development Negative regulation of neurogenesis Glial cell differentiation Response to mechanical stimulus Regulation of neuron differentiation Extracellular matrix organization Extracellular structure organization Positive regulation of neuroblast proliferation |
| Dividing Radial Glia | UHRF1, CTB-175P5.4, RP11-138A9.1, RP11-143K11.1, AC004447.2, SNORA59B, CTC-503J8.6, RP11-138A9.2, RP11-95D17.1, THAP9-AS1, SNHG1, CTD-2017D11.1, RP11-58B17.2, DYNLL1-AS1 | MKI67, KIF15, CCNB2, CDK1, UBE2C, FAM64A, NDC80, AURKB, MELK, TPX2, CDCA5, HIST1H1B, BIRC5, ZWINT, TOP2A | Mitotic cell cycle Nuclear division Organelle fission Mitotic nuclear division Chromosome segregation Regulation of cell cycle process Cell cycle checkpoint Chromosome organization DNA repair Microtubule-based process |
| Intermediate Progenitor | LINC-TMEM200C-1, RP11-798G7.8, RP11-35IJ23.1-AS1, RP3-326L13.3, CTD-2245E15.3, C1orf132, AC084018.1, RP11-73O6.3, RP11-594N15.3, RP11-436D23.1, AC0838848.8, DGCR11, RP11-456K23.1, RP6-24A23.3, RP1-20C7.6 | PPP1R17, EOMES, NHLH1, SSTR2, SETD7, CCDC129, SIPA1L2, NPR3, FAM60A, SLCO4C1, TRIM45, INHBB, UBL7, STX8, TMEM206 | Dicarboxylic acid biosynthetic process Glutamine family amino acid biosynthetic process GPI anchor metabolic process Regulation of triglyceride biosynthetic process Glutamate metabolic process Neuroblast proliferation Neuromuscular synaptic transmission GPI anchor biosynthetic process Positive regulation of triglyceride metabolic process Positive regulation of triglyceride biosynthetic process |
| New Neuron | RP5-1024G6.8, LINC-PTCHD2-3, RP11-513M16.8, RP11-661O13.1, RP11-524C21.2, RP11-356K23.1, LINC01105 | SLC24A2, NRP1, RASGEF1, PALMD, SEMA3C, KCNQ3, UNC5D, SLC17A6, DOK6, SEZ6, DCC, SORBS2, FAM126A, ZNF804A, PPP2R2B | Limb bud formation Cardiac ventricle morphogenesis Cardiac chamber morphogenesis Axon extension Regulation of neuron differentiation Neuron projection extension Positive regulation of neuron differentiation Positive regulation of neurogenesis Glial cell development Regulation of neuron projection development |
| Maturing Neuron | MIR137HG, LINC00599, PWAR6, SIK3-IT1, RP11-53O19.3, RP11-402L6.1, RP11-18I14.10, RP11-486F17.1, NAV2-AS3, DAPK1-IT1, RP11-397O4.1, RP11-64K12.10, LINC00643, RP3-462E2.5, LINC-TMEM182-5 | SLC44A5, GRIN2B, CCBE1, CDH13, CAMK2B, SATB2, ARPP21, ADRA2A, DAB1, GLRA2, GPR85, KIAA0319, MCTP1, ADCY1, FLRT2 | Limb bud formation Cardiac ventricle morphogenesis Cardiac chamber morphogenesis Axon extension Regulation of neuron differentiation Neuron projection extension Positive regulation of neuron differentiation Positive regulation of neurogenesis Glial cell development Regulation of neuron projection development |
| Interneuron | DLX-AS1, RP11-588P7.1, SOX2-OT, GS1-18A18.1, MEG3, LINC-DKFZP761K2322-2, GRIP2, AC87393.1, LINC00966, RP11-450H6.3, RP13-514E23.1, RP11-379H18.1, RP11-69E11.4, AC012358.8, LINC-TBCC-1 | ERBB4, GAD1, MAF, DLX2, NRXN3, FAM65B, DLX5, PLS3, PDZRN3, LHX6, DLX6, THRB, SCGN, IGF1, CELF4 | GABA synthesis, release, reuptake and degradation Transmission across chemical synapses Neurotransmitter release cycle Nuclear receptor transcription pathway Signaling by ERBB2 Signaling by FGFR Signaling by FGFR in disease Neuronal system Downstream signal transduction Downstream signaling of activated FGFR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeSouza, P.A.; Qu, X.; Chen, H.; Patel, B.; Maher, C.A.; Kim, A.H. Long, Noncoding RNA Dysregulation in Glioblastoma. Cancers 2021, 13, 1604. https://doi.org/10.3390/cancers13071604
DeSouza PA, Qu X, Chen H, Patel B, Maher CA, Kim AH. Long, Noncoding RNA Dysregulation in Glioblastoma. Cancers. 2021; 13(7):1604. https://doi.org/10.3390/cancers13071604
Chicago/Turabian StyleDeSouza, Patrick A., Xuan Qu, Hao Chen, Bhuvic Patel, Christopher A. Maher, and Albert H. Kim. 2021. "Long, Noncoding RNA Dysregulation in Glioblastoma" Cancers 13, no. 7: 1604. https://doi.org/10.3390/cancers13071604
APA StyleDeSouza, P. A., Qu, X., Chen, H., Patel, B., Maher, C. A., & Kim, A. H. (2021). Long, Noncoding RNA Dysregulation in Glioblastoma. Cancers, 13(7), 1604. https://doi.org/10.3390/cancers13071604