
Assessment of Immunological Features in Muscle-Invasive Bladder Cancer Prognosis Using Ensemble Learning

 , , ,
, , ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Multiplex Immunofluorescence and Whole Slide Imaging
2.3. Detection of Cell Nuclei
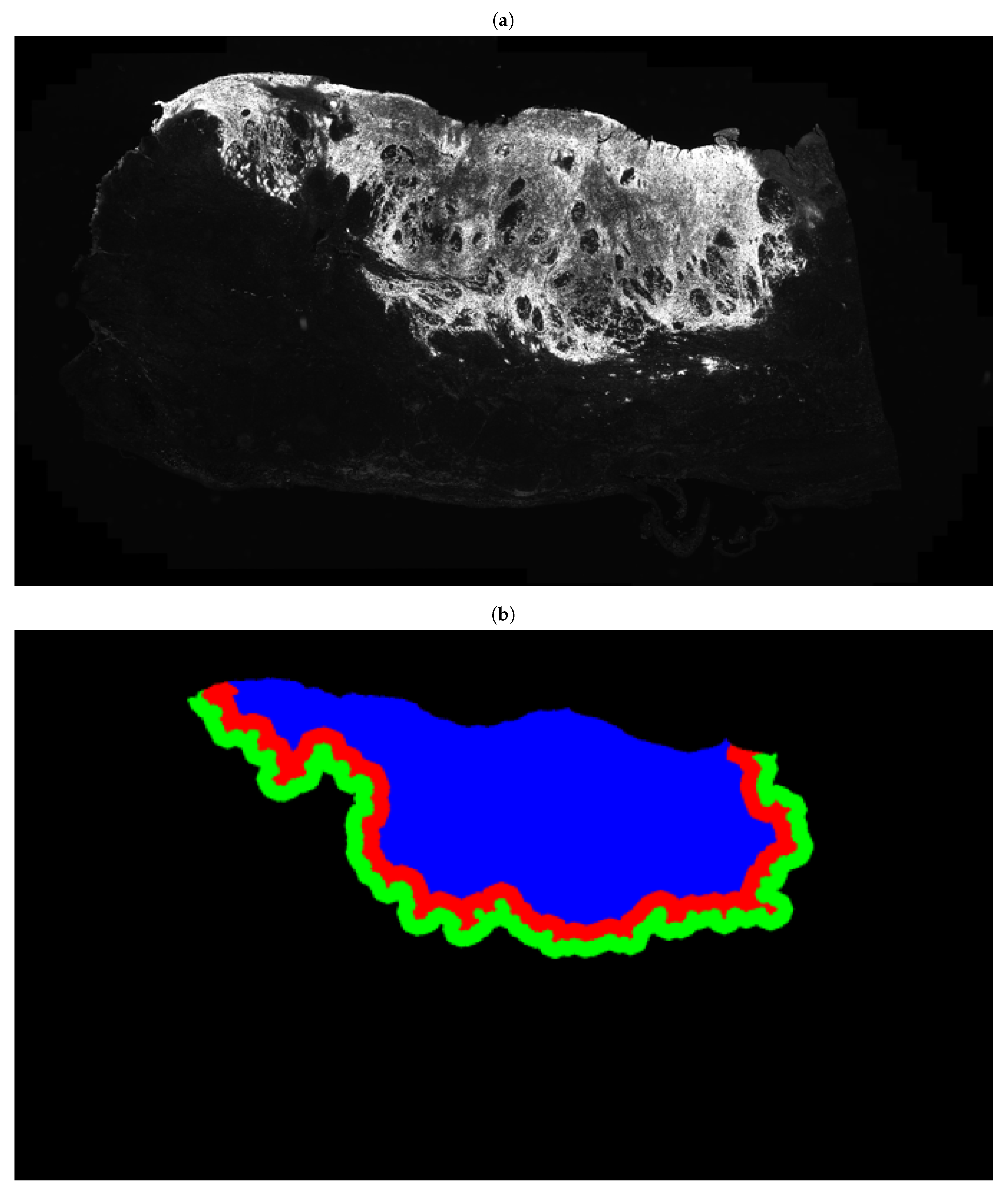
2.4. Segmentation of Epithelial Cells for the Identification of Tumour Buds
2.5. Cell Classification
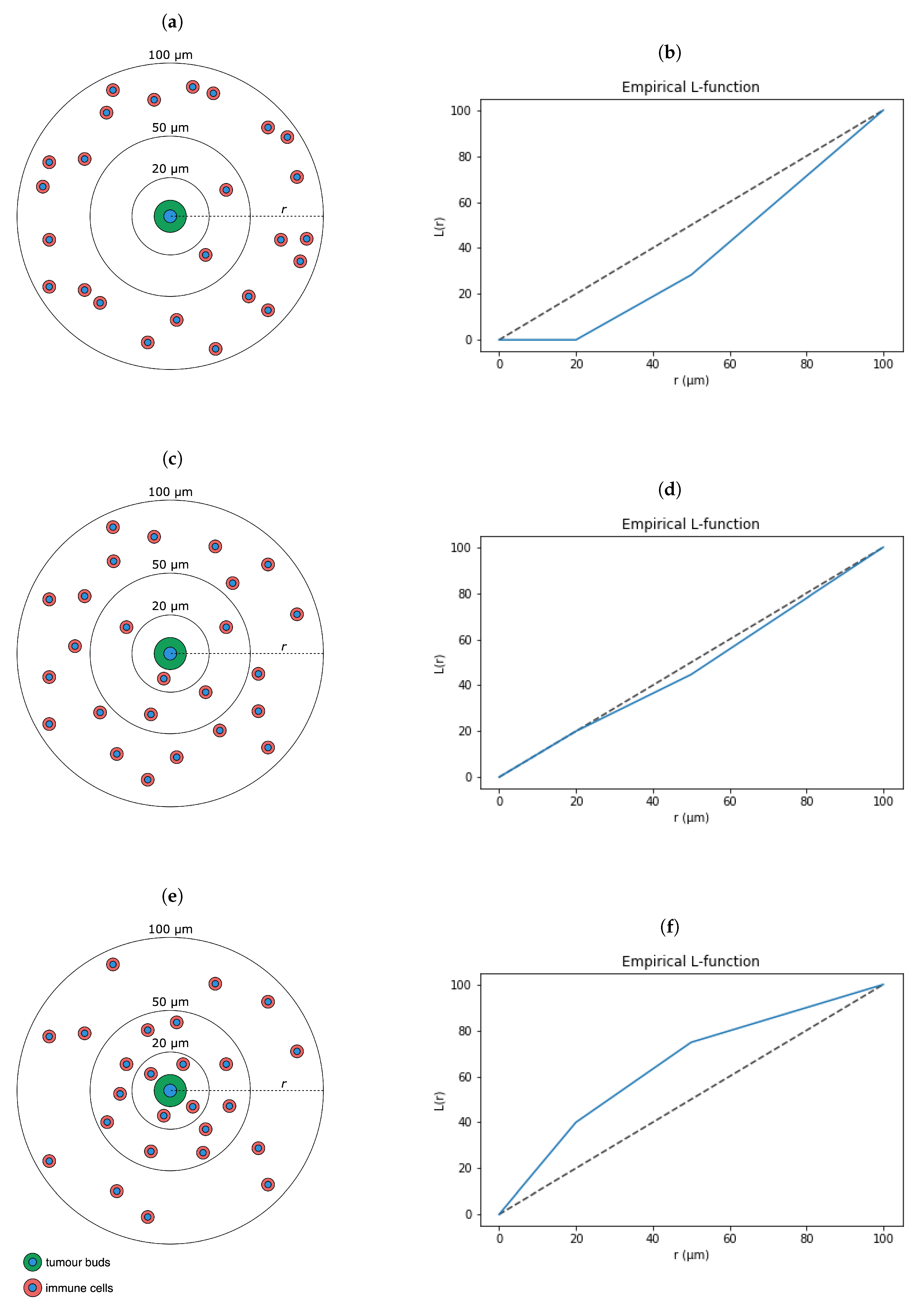
2.6. Pairwise Spatial Distributions of Lymphocytes, Macrophages, Tumour Buds and PD-L1
2.7. Binary Survival Analysis
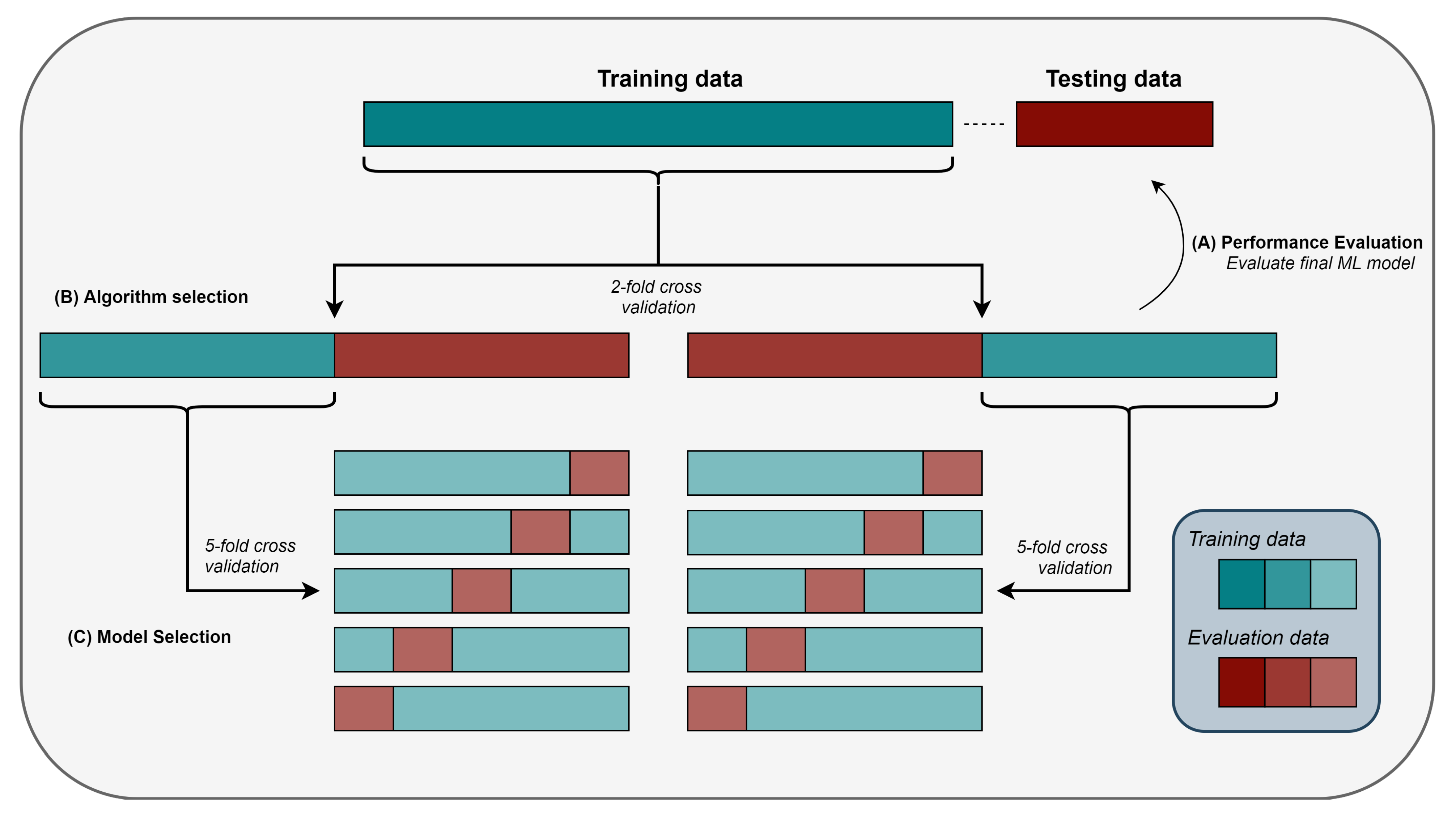
2.8. Model Selection, Algorithm Selection, and Performance Evaluation
2.9. Stratified Sampling
3. Results
3.1. Patient Characteristics
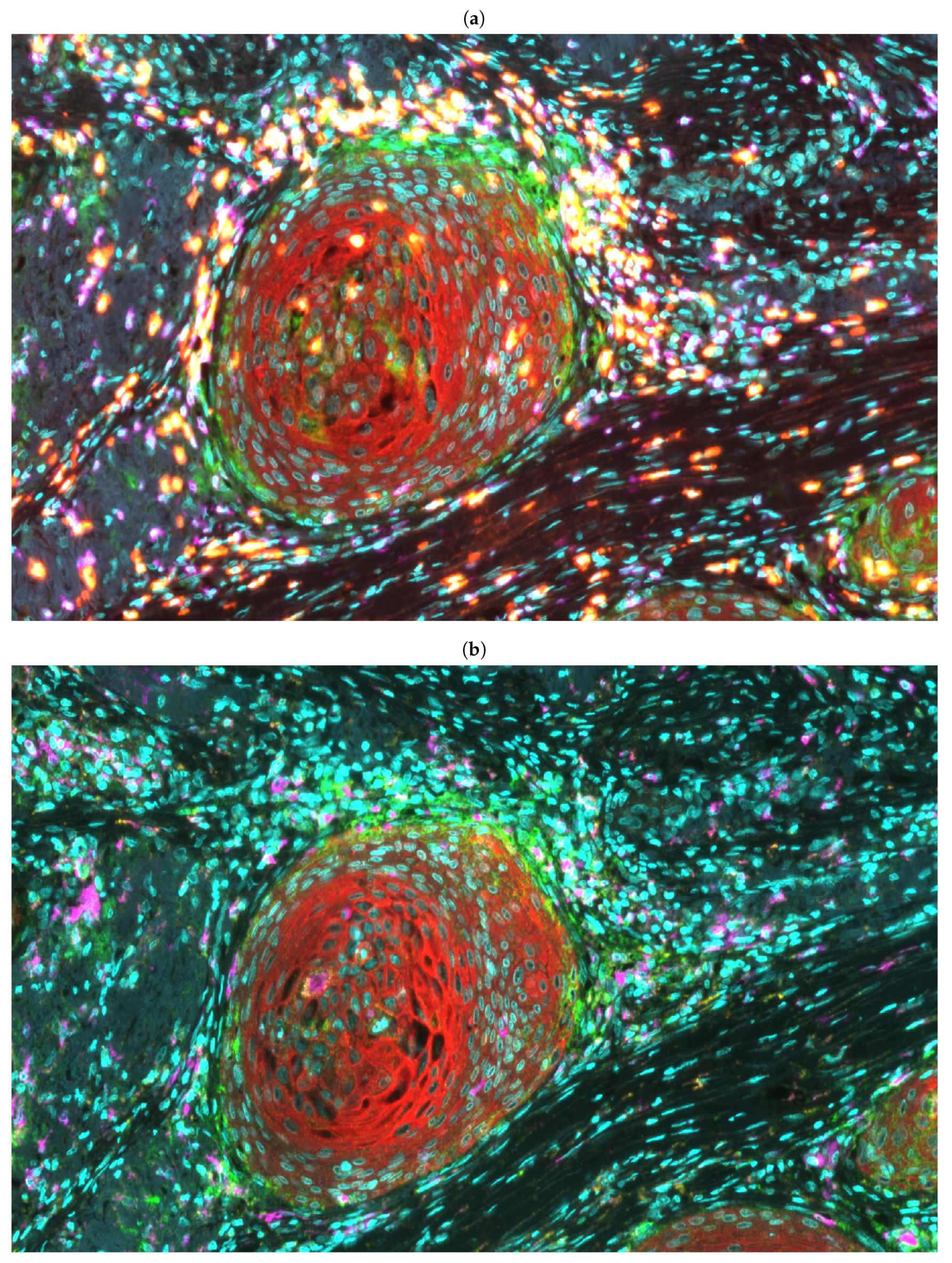
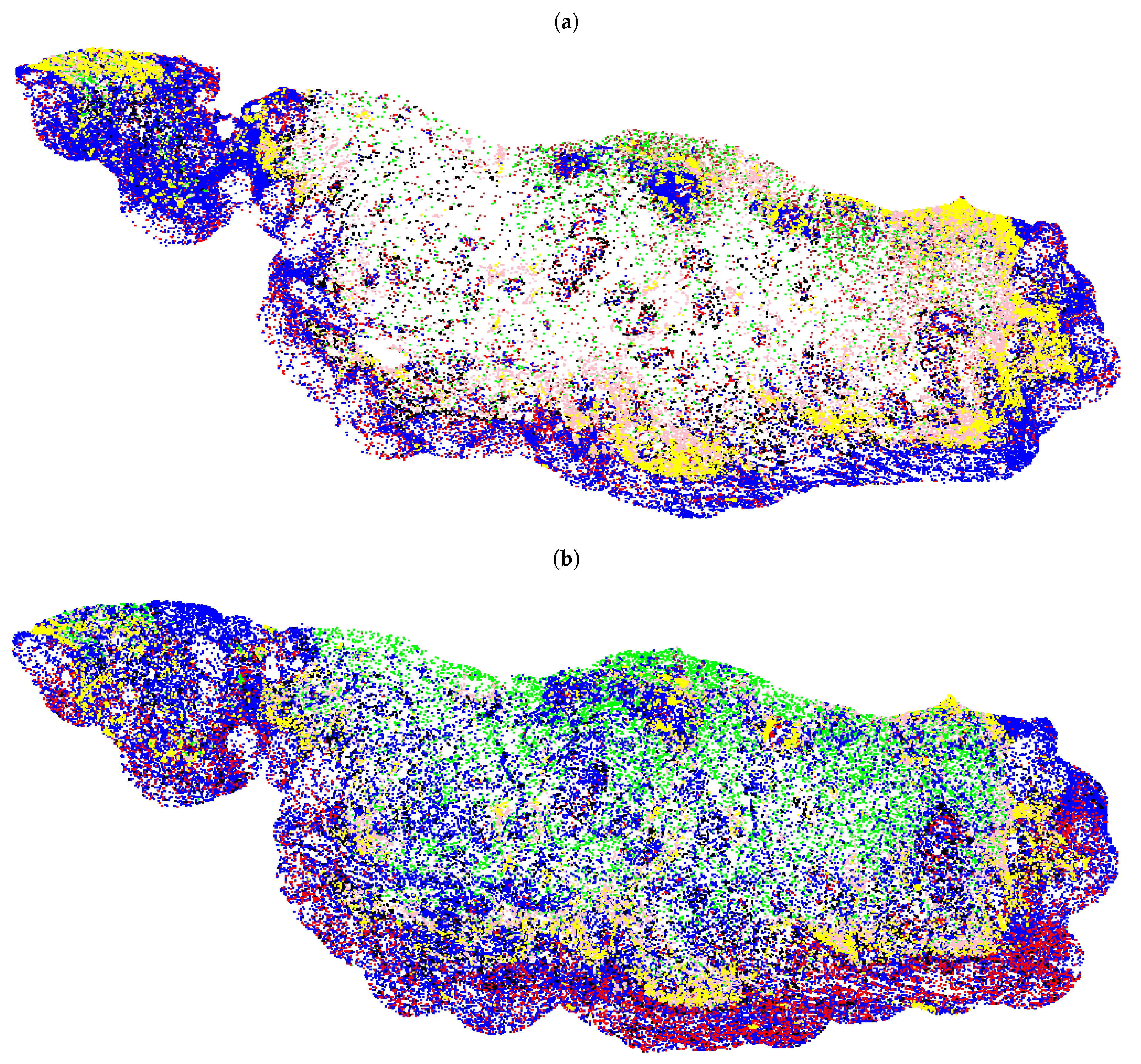
3.2. Fully Automated Feature Extraction
3.3. Feature Space and Feature Selection
3.4. Machine Learning Models and Optimizing Metric
3.5. Proposed Ensemble Model
3.6. Pessimistic Bias
3.7. Comparing against TNM Staging
3.8. Post-hoc Analysis of Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Frantzi, M.; Van Kessel, K.E.; Zwarthoff, E.C.; Marquez, M.; Rava, M.; Malats, N.; Merseburger, A.S.; Katafigiotis, I.; Stravodimos, K.; Mullen, W.; et al. Development and validation of urine-based peptide biomarker panels for detecting bladder cancer in a multi-center study. Clin. Cancer Res. 2016, 22, 4077–4086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.M.; DeCastro, G.J.; Steinberg, G.D. Urothelial carcinoma of the bladder: Definition, treatment and future efforts. Nat. Rev. Urol. 2011, 8, 631. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017, 171, 540–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, A.P.; Fantini, D.; Shilatifard, A.; Schaeffer, E.M.; Meeks, J.J. The evolving genomic landscape of urothelial carcinoma. Nat. Rev. Urol. 2017, 14, 215. [Google Scholar] [CrossRef]
- American Joint Committee on Cancer. AJCC—Cancer Staging Manual. Available online: https://cancerstaging.org/references-tools/deskreferences/Pages/default.aspx (accessed on 10 October 2020).
- Alifrangis, C.; McGovern, U.; Freeman, A.; Powles, T.; Linch, M. Molecular and histopathology directed therapy for advanced bladder cancer. Nat. Rev. Urol. 2019, 16, 465–483. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346. [Google Scholar] [CrossRef]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Go, S.I.; Song, D.H.; Park, S.W.; Kim, H.R.; Jang, I.; Kim, J.D.; Lee, J.S.; Lee, G.W. Prognostic impact of CD8 and programmed death-ligand 1 expression in patients with resectable non-small cell lung cancer. Br. J. Cancer 2019, 120, 547. [Google Scholar] [CrossRef] [Green Version]
- Foerster, F.; Hess, M.; Gerhold-Ay, A.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Binder, H.; Bockamp, E. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci. Rep. 2018, 8, 5351. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914. [Google Scholar] [CrossRef]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451. [Google Scholar] [CrossRef] [Green Version]
- Lohneis, P.; Sinn, M.; Klein, F.; Bischoff, S.; Striefler, J.K.; Wislocka, L.; Sinn, B.V.; Pelzer, U.; Oettle, H.; Riess, H.; et al. Tumour buds determine prognosis in resected pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 1485. [Google Scholar] [CrossRef]
- Li, L.; Sun, R.; Miao, Y.; Tran, T.; Adams, L.; Roscoe, N.; Xu, B.; Manyam, G.C.; Tan, X.; Zhang, H.; et al. PD-1/PD-L1 expression and interaction by automated quantitative immunofluorescent analysis show adverse prognostic impact in patients with diffuse large B-cell lymphoma having T-cell infiltration: A study from the International DLBCL Consortium Program. Mod. Pathol. 2019, 32, 741–754. [Google Scholar] [CrossRef]
- Fumet, J.D.; Richard, C.; Ledys, F.; Klopfenstein, Q.; Joubert, P.; Routy, B.; Truntzer, C.; Gagné, A.; Hamel, M.A.; Guimaraes, C.F.; et al. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer 2018, 119, 950. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Tołoczko, A.; Hein, A.; Bouligny, A.L.; et al. Dose-response association of CD8+ tumor-infiltrating lymphocytes and survival time in high-grade serous ovarian cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Lugli, A. Tumour budding in colorectal cancer: Molecular rationale for clinical translation. Nat. Rev. Cancer 2018, 18, 203–204. [Google Scholar] [CrossRef]
- Ueno, H.; Murphy, J.; Jass, J.; Mochizuki, H.; Talbot, I. Tumour budding as an index to estimate the potential of aggressiveness in rectal cancer. Histopathology 2002, 40, 127–132. [Google Scholar] [CrossRef]
- Gujam, F.; McMillan, D.; Mohammed, Z.; Edwards, J.; Going, J. The relationship between tumour budding, the tumour microenvironment and survival in patients with invasive ductal breast cancer. Br. J. Cancer 2015, 113, 1066. [Google Scholar] [CrossRef] [Green Version]
- Lugli, A.; Kirsch, R.; Ajioka, Y.; Bosman, F.; Cathomas, G.; Dawson, H.; El Zimaity, H.; Fléjou, J.F.; Hansen, T.P.; Hartmann, A.; et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod. Pathol. 2017, 30, 1299. [Google Scholar] [CrossRef]
- Brieu, N.; Gavriel, C.G.; Nearchou, I.P.; Harrison, D.J.; Schmidt, G.; Caie, P.D. Automated tumour budding quantification by machine learning augments TNM staging in muscle-invasive bladder cancer prognosis. Sci. Rep. 2019, 9, 5174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nearchou, I.P.; Lillard, K.; Gavriel, C.G.; Ueno, H.; Harrison, D.J.; Caie, P.D. Automated analysis of lymphocytic infiltration, tumor budding, and their spatial relationship improves prognostic accuracy in colorectal cancer. Cancer Immunol. Res. 2019, 7, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and function of the PD-L1 checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Zhang, C.; Berry, G.J.; Altman, R.B.; Ré, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Madabhushi, A.; Lee, G. Image analysis and machine learning in digital pathology: Challenges and opportunities. Med. Image Anal. 2016, 33, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Dimitriou, N.; Arandjelović, O.; Harrison, D.J.; Caie, P.D. A principled machine learning framework improves accuracy of stage II colorectal cancer prognosis. NPJ Digit. Med. 2018, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mokarram, R.; Emadi, M. Classification in Non-linear Survival Models Using Cox Regression and Decision Tree. Ann. Data Sci. 2017, 4, 329–340. [Google Scholar] [CrossRef]
- Wang, P.; Li, Y.; Reddy, C.K. Machine Learning for Survival Analysis: A Survey. ACM Comput. Surv. 2017, 51, 1–36. [Google Scholar] [CrossRef]
- Brieu, N.; Schmidt, G. Learning size adaptive local maxima selection for robust nuclei detection in histopathology images. In Proceedings of the 2017 IEEE 14th International Symposium on Biomedical Imaging (ISBI 2017), Melbourne, Australia, 18–21 April 2017; pp. 937–941. [Google Scholar]
- Criminisi, A.; Shotton, J.; Bucciarelli, S. Decision forests with long-range spatial context for organ localization in CT volumes. In Proceedings of the Medical Image Computing and Computer-Assisted Intervention (MICCAI), London, UK, 20–24 September 2009; pp. 69–80. [Google Scholar]
- Brieu, N.; Pauly, O.; Zimmermann, J.; Binnig, G.; Schmidt, G. Slide-specific models for segmentation of differently stained digital histopathology whole slide images. In Proceedings of the Medical Imaging 2016: Image Processing, San Diego, CA, USA, 1–3 March 2016; Volume 9784, p. 978410. [Google Scholar]
- Brieu, N.; Gavriel, C.G.; Harrison, D.J.; Caie, P.D.; Schmidt, G. Context-based interpolation of coarse deep learning prediction maps for the segmentation of fine structures in immunofluorescence images. In Proceedings of the Medical Imaging 2018: Digital Pathology, Pathology, Houston, Texas, USA, 6 March 2018; International Society for Optics and Photonics: Bellingham, WA, USA, 2018; Volume 10581, p. 105810. [Google Scholar]
- Ronneberger, O.; Fischer, P.; Brox, T. U-net: Convolutional networks for biomedical image segmentation. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Munich, Germany, 5–9 October 2015; pp. 234–241. [Google Scholar]
- Ripley, B.D. Modelling spatial patterns. J. R. Stat. Soc. Ser. B (Methodol.) 1977, 39, 172–192. [Google Scholar] [CrossRef]
- Besag, J. Contribution to the discussion on Dr Ripley’s paper. JR Stat. Soc. 1977, 39, 193–195. [Google Scholar]
- Carstens, J.L.; De Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Leung, K.; Elashoff, M.R.; Afifi, A.A. Censoring Issues In Survival Analysis. Annu. Rev. Public Health 1997, 18, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Kemi, N.; Eskuri, M.; Kauppila, J.H. Tumour-stroma ratio and 5-year mortality in gastric adenocarcinoma: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Noon, A.; Albertsen, P.; Thomas, F.; Rosario, D.; Catto, J. Competing mortality in patients diagnosed with bladder cancer: Evidence of undertreatment in the elderly and female patients. Br. J. Cancer 2013, 108, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Bergstra, J.; Yamins, D.; Cox, D.D. Making a Science of Model Search: Hyperparameter Optimization in Hundreds of Dimensions for Vision Architectures. In Proceedings of the 30th International Conference on International Conference on Machine Learning, Atlanta, GA, USA, 16–21 June 2013; Volume 28, pp. I-115–I-123. [Google Scholar]
- Bergstra, J.; Bengio, Y. Random Search for Hyper-parameter Optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- Wolpert, D.H.; Macready, W.G. No Free Lunch Theorems for Optimization. Trans. Evol. Comp 1997, 1, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Harder, N.; Athelogou, M.; Hessel, H.; Brieu, N.; Yigitsoy, M.; Zimmermann, J.; Baatz, M.; Buchner, A.; Stief, C.G.; Kirchner, T.; et al. Tissue Phenomics for prognostic biomarker discovery in low-and intermediate-risk prostate cancer. Sci. Rep. 2018, 8, 4470. [Google Scholar] [CrossRef]
- Binnig, G.; Huss, R.; Schmidt, G. Tissue Phenomics: Profiling Cancer Patients for Treatment Decisions; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Athelogou, M.; Schmidt, G.; Schäpe, A.; Baatz, M.; Binnig, G. Cognition network technology–a novel multimodal image analysis technique for automatic identification and quantification of biological image contents. In Imaging Cellular and Molecular Biological Functions; Springer: Berlin/Heidelberg, Germany, 2007; pp. 407–422. [Google Scholar]
- Raschka, S. Model Evaluation, Model Selection, and Algorithm Selection in Machine Learning. arXiv 2018, arXiv:1811.12808. [Google Scholar]
- Louppe, G.; Wehenkel, L.; Sutera, A.; Geurts, P. Understanding variable importances in forests of randomized trees. In Advances in Neural Information Processing Systems 26; Burges, C.J.C., Bottou, L., Welling, M., Ghahramani, Z., Weinberger, K.Q., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2013; pp. 431–439. [Google Scholar]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93. [Google Scholar] [CrossRef] [Green Version]
- Yagi, T.; Baba, Y.; Okadome, K.; Kiyozumi, Y.; Hiyoshi, Y.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Watanabe, M.; et al. Tumour-associated macrophages are associated with poor prognosis and programmed death ligand 1 expression in oesophageal cancer. Eur. J. Cancer 2019, 111, 38–49. [Google Scholar] [CrossRef]
- Keller, M.D.; Neppl, C.; Irmak, Y.; Hall, S.R.; Schmid, R.A.; Langer, R.; Berezowska, S. Adverse prognostic value of PD-L1 expression in primary resected pulmonary squamous cell carcinomas and paired mediastinal lymph node metastases. Mod. Pathol. 2018, 31, 101. [Google Scholar] [CrossRef]
- Masugi, Y.; Abe, T.; Ueno, A.; Fujii-Nishimura, Y.; Ojima, H.; Endo, Y.; Fujita, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; et al. Characterization of spatial distribution of tumor-infiltrating CD8+ T cells refines their prognostic utility for pancreatic cancer survival. Mod. Pathol. 2019, 32, 1495–1507. [Google Scholar] [CrossRef]
- Xue, S.; Song, G.; Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef] [Green Version]
- Caie, P.; Dimitriou, N.; Nearchou, I.; Arandjelovic, O.; Harrison, D. Artificial Intelligence Driving Automated Pathology: ICAIRD and Beyond. Virchows Arch. 2019, 475, S60. [Google Scholar]
- Parra, E.R.; Francisco-Cruz, A.; Wistuba, I.I. State-of-the-art of profiling immune contexture in the era of multiplexed staining and digital analysis to study paraffin tumor tissues. Cancers 2019, 11, 247. [Google Scholar] [CrossRef] [Green Version]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. Elife 2018, 7, e36967. [Google Scholar] [CrossRef]
- König, L.; Mairinger, F.D.; Hoffmann, O.; Bittner, A.K.; Schmid, K.W.; Kimmig, R.; Kasimir-Bauer, S.; Bankfalvi, A. Dissimilar patterns of tumor-infiltrating immune cells at the invasive tumor front and tumor center are associated with response to neoadjuvant chemotherapy in primary breast cancer. BMC Cancer 2019, 19, 120. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef]
- Riihijärvi, S.; Fiskvik, I.; Taskinen, M.; Vajavaara, H.; Tikkala, M.; Yri, O.; Karjalainen-Lindsberg, M.L.; Delabie, J.; Smeland, E.; Holte, H.; et al. Prognostic influence of macrophages in patients with diffuse large B-cell lymphoma: A correlative study from a Nordic phase II trial. Haematologica 2015, 100, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Chaput, N.; Svrcek, M.; Aupérin, A.; Locher, C.; Drusch, F.; Malka, D.; Taïeb, J.; Goéré, D.; Ducreux, M.; Boige, V. Tumour-infiltrating CD68+ and CD57+ cells predict patient outcome in stage II–III colorectal cancer. Br. J. Cancer 2013, 109, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
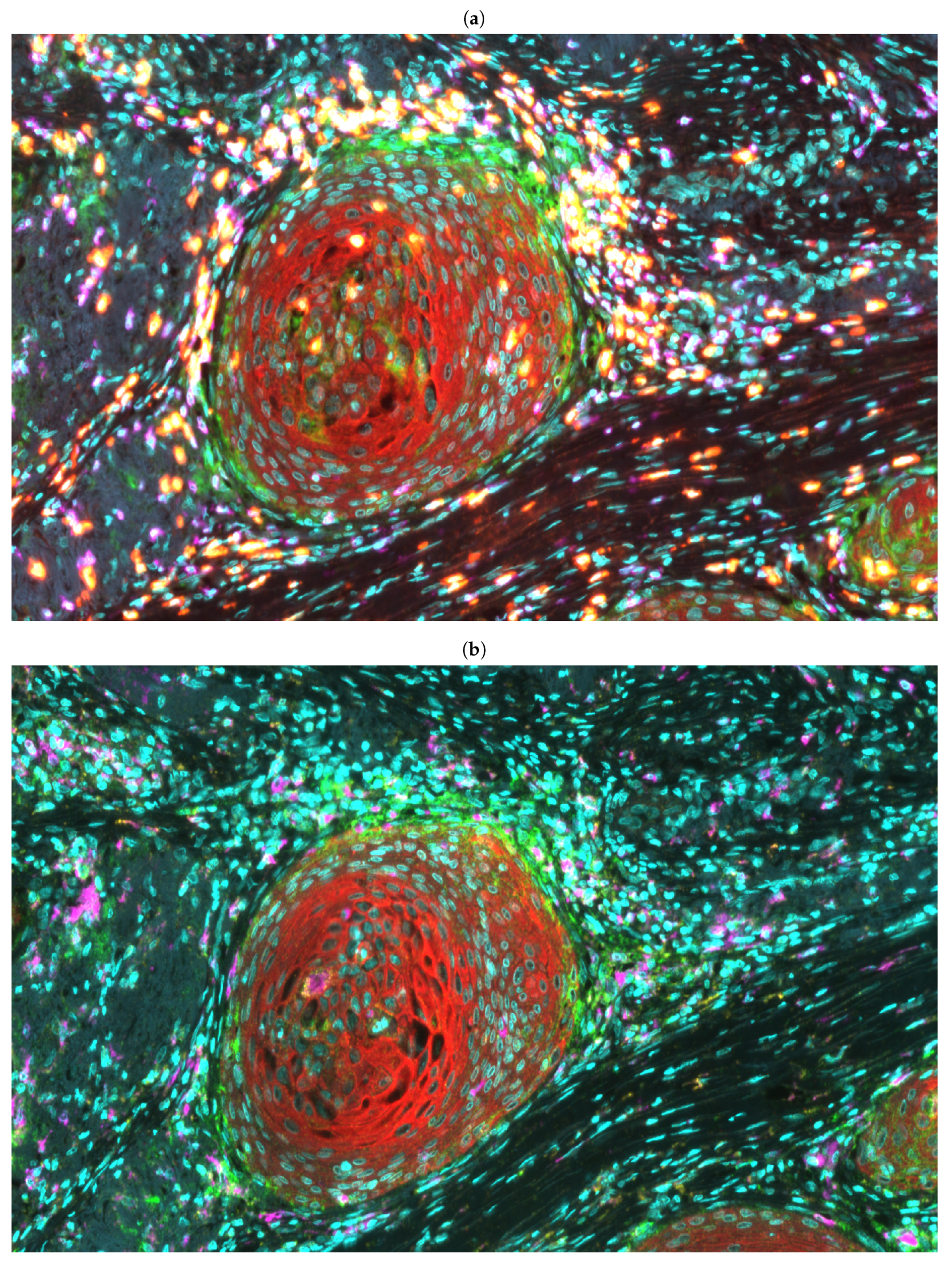






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Summary |
|---|---|
| MIBC patients | N = 78 |
| Median survival (range) | 19 (1–113) months |
| Age | years |
| Gender | Male; Female |
| TNM stage | |
| II | 17 (22%) |
| IIIA | 29 (37%) |
| IIIB | 5 (6%) |
| IV | 27 (35%) |
| Tumour (T) | |
| T2 | 18 (23%) |
| T3 | 39 (50%) |
| T4 | 21 (27%) |
| Node (N) | |
| N0 | 57 (73%) |
| N1 | 13 (17%) |
| N2 | 8 (10%) |
| Metastasis (M) | |
| M0 | 51 (65%) |
| M1 | 27 (35%) |
| Training Set | Testing Set | |||
|---|---|---|---|---|
| Evaluation Metrics | Ensemble Model | TNM Staging | Ensemble Model | TNM Staging |
| AUROC | 98.3 | |||
| Accuracy | ||||
| Sensitivity | ||||
| Specificity | ||||
| F1 score | ||||
| Hazard ratio | ||||
| (, ) * | (, ) * | (, ) * | (, ) * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavriel, C.G.; Dimitriou, N.; Brieu, N.; Nearchou, I.P.; Arandjelović, O.; Schmidt, G.; Harrison, D.J.; Caie, P.D. Assessment of Immunological Features in Muscle-Invasive Bladder Cancer Prognosis Using Ensemble Learning. Cancers 2021, 13, 1624. https://doi.org/10.3390/cancers13071624
Gavriel CG, Dimitriou N, Brieu N, Nearchou IP, Arandjelović O, Schmidt G, Harrison DJ, Caie PD. Assessment of Immunological Features in Muscle-Invasive Bladder Cancer Prognosis Using Ensemble Learning. Cancers. 2021; 13(7):1624. https://doi.org/10.3390/cancers13071624
Chicago/Turabian StyleGavriel, Christos G., Neofytos Dimitriou, Nicolas Brieu, Ines P. Nearchou, Ognjen Arandjelović, Günter Schmidt, David J. Harrison, and Peter D. Caie. 2021. "Assessment of Immunological Features in Muscle-Invasive Bladder Cancer Prognosis Using Ensemble Learning" Cancers 13, no. 7: 1624. https://doi.org/10.3390/cancers13071624
APA StyleGavriel, C. G., Dimitriou, N., Brieu, N., Nearchou, I. P., Arandjelović, O., Schmidt, G., Harrison, D. J., & Caie, P. D. (2021). Assessment of Immunological Features in Muscle-Invasive Bladder Cancer Prognosis Using Ensemble Learning. Cancers, 13(7), 1624. https://doi.org/10.3390/cancers13071624