
Role for the Histone Demethylase KDM4B in Rhabdomyosarcoma via CDK6 and CCNA2: Compensation by KDM4A and Apoptotic Response of Targeting Both KDM4B and KDM4A

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNA Screen and Transfections
2.3. Drug Sensitivity Analysis
2.4. Immunofluorescence
2.5. Immunohistochemistry
2.6. KDM4B Gene Expression
2.7. Cell Proliferation, Cell Cycle and Apoptosis Assays
2.8. RNA-Sequencing and Bioinformatics
2.9. Western Blotting
2.10. Chromatin Immunoprecipitation and qPCR
2.11. shRNA Transfections
2.12. Statistics
3. Results
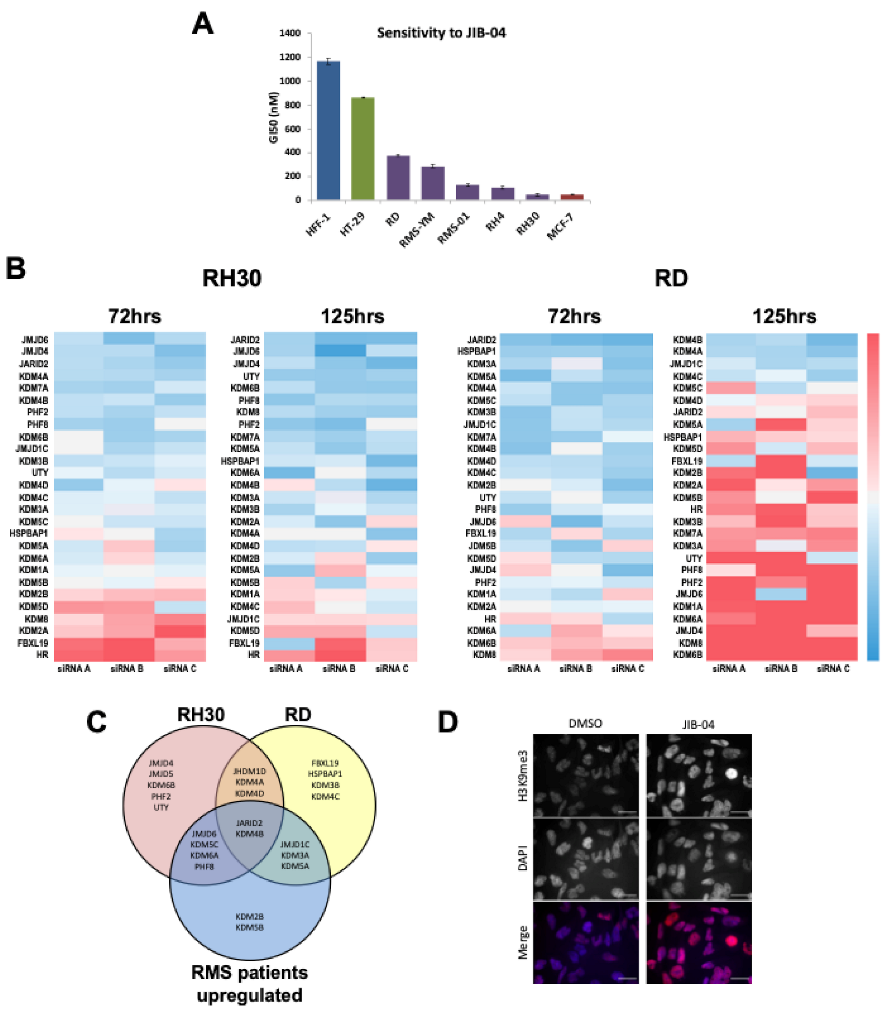
3.1. HDMs Are Important Mediators of Proliferation In Rhabdomyosarcomas
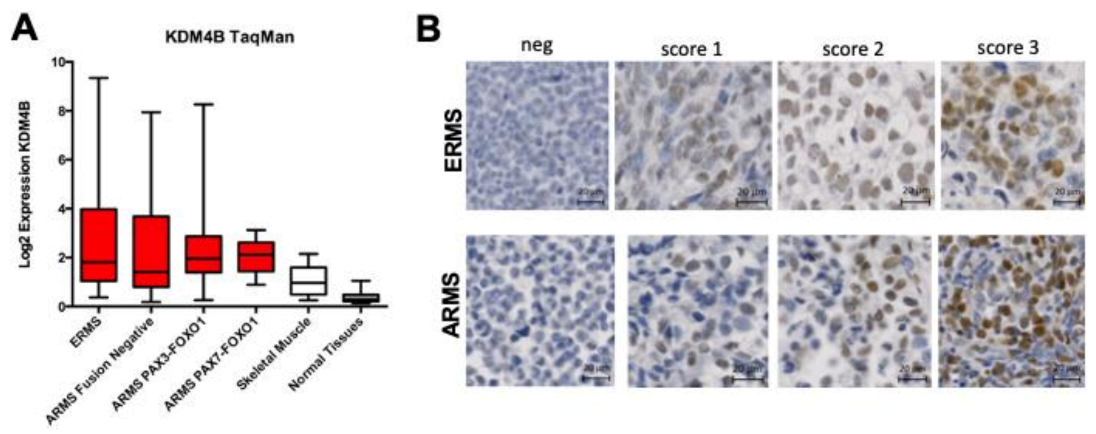
3.2. KDM4B Is Highly Expressed and Correlates with A Proliferative Marker in RMS
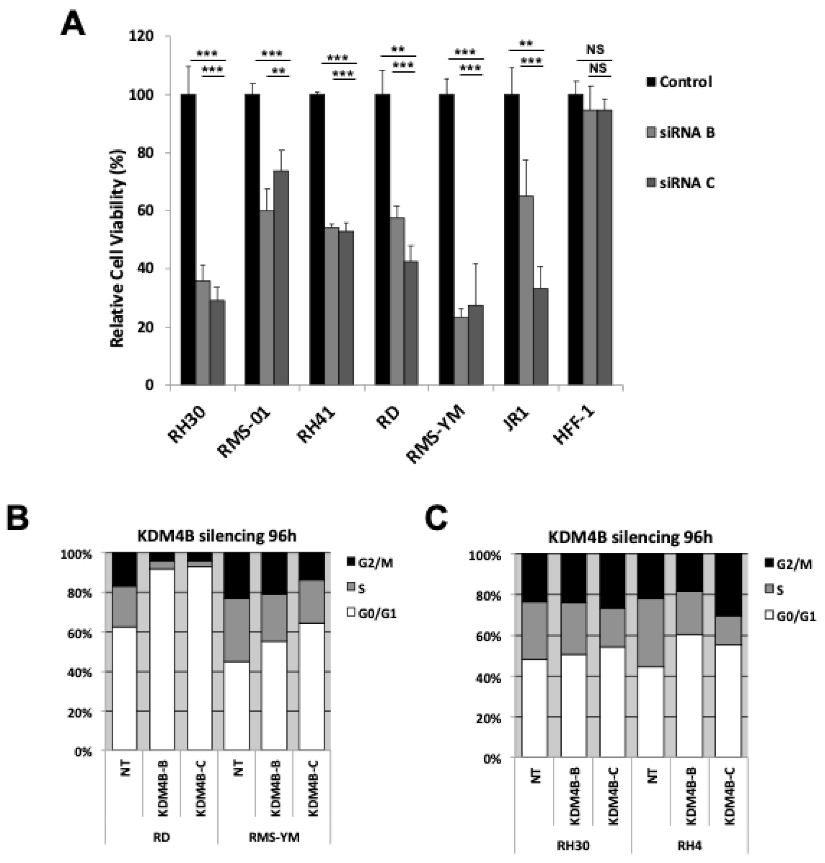
3.3. KDM4B Silencing Results in A Reduction in Proliferation in RMS cells
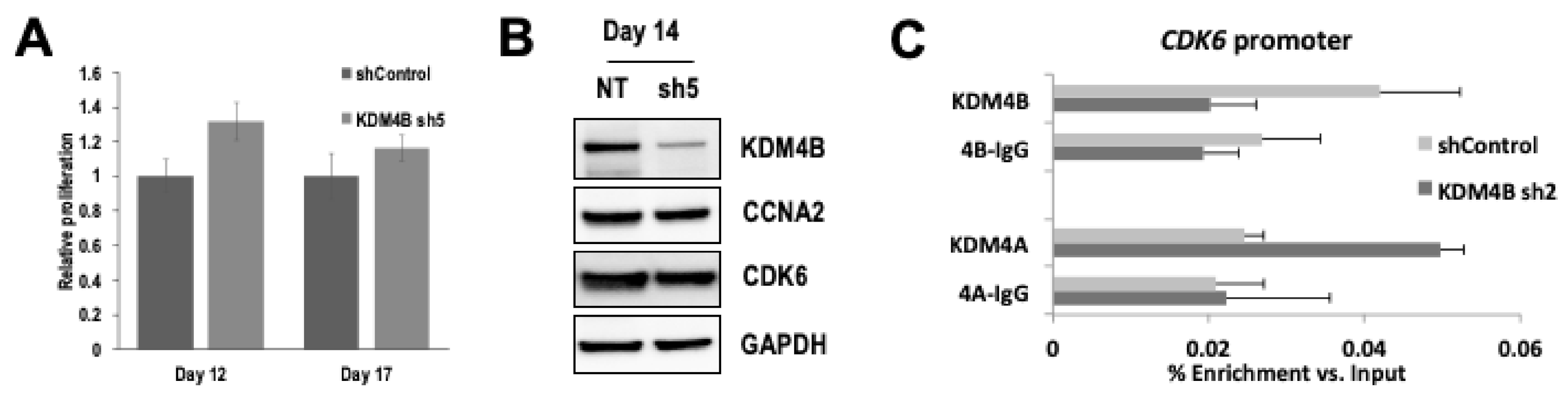
3.4. KDM4B Regulates Transcription of G1/S and G2/M Checkpoint Genes
3.5. Sustained KDM4B Silencing Leads to Resistance to Growth Suppression via Molecular Compensation by KDM4A
3.6. Dual Silencing of KDM4A and KDM4B Results in Apoptosis in RMS Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; Angeles, A.D.L.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and Combinatorial Functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in Mouse Embryonic Stem Cell Identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.T.; Kooistra, S.M.; Radzisheuskaya, A.; Laugesen, A.; Johansen, J.V.; Hayward, D.G.; Nilsson, J.; Agger, K.; Helin, K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016, 35, 1550–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Bollig-Fischer, A.; Kreike, B.; van de Vijver, M.J.; Abrams, J.; Ethier, S.P.; Yang, Z.Q. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 2009, 28, 4491–4500. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.-H.; Zhang, W.; Chen, X.; George, J.; Ng, H.-H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007, 21, 2545–2557. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wary, K.K.; Revskoy, S.; Gao, X.; Tsang, K.; Komarova, Y.A.; Rehman, J.; Malik, A.B. Histone Demethylases KDM4A and KDM4C Regulate Differentiation of Embryonic Stem Cells to Endothelial Cells. Stem Cell Rep. 2015, 5, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Labbé, R.M.; Holowatyj, A.; Yang, Z.-Q. Histone lysine demethylase (KDM) subfamily 4: Structures, functions and therapeutic potential. Am. J. Transl. Res. 2013, 6, 1–15. [Google Scholar]
- Young, L.C.; Hendzel, M.J. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem. Cell Biol. 2013, 91, 369–377. [Google Scholar] [CrossRef]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 Histone Demethylases: Epigenetic Regulators in Cancer Cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Guo, W.; Shen, J.K.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Rhabdomyosarcoma: Advances in Molecular and Cellular Biology. Sarcoma. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ciarapica, R.; Carcarino, E.; Adesso, L.; De Salvo, M.; Bracaglia, G.; Leoncini, P.P.; Dall’Agnese, A.; Verginelli, F.; Milano, G.M.; Boldrini, R.; et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 2014, 14, 139. [Google Scholar] [CrossRef] [Green Version]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, E.; Crose, L.E.S.; Linardic, C.M.; Safe, S. Histone Deacetylase Inhibitors Inhibit Rhabdomyosarcoma by Reactive Oxygen Species–Dependent Targeting of Specificity Protein Transcription Factors. Mol. Cancer Ther. 2015, 14, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-H.; Jothi, M.; Gudkov, A.V.; Mal, A.K. Histone Methyltransferase KMT1A Restrains Entry of Alveolar Rhabdomyosarcoma Cells into a Myogenic Differentiated State. Cancer Res. 2011, 71, 3921–3931. [Google Scholar] [CrossRef] [Green Version]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.T.; Kumar, S.; Best, A.M.; Ruiz, J.A.; Bruick, R.K.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor Activity of Sustained N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Williamson, D.; Missiaglia, E.; Pritchard-Jones, K.; Oberlin, O.; Shipley, J.M.; Delattre, O.; De Reyniès, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; et al. Fusion Gene-Negative Alveolar Rhabdomyosarcoma is Clinically and Molecularly Indistinguishable from Embryonal Rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef] [Green Version]
- Toyokawa, G.; Cho, H.-S.; Iwai, Y.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Shimizu, N.; Tanaka, H.; Tsunoda, T.; et al. The Histone Demethylase JMJD2B Plays an Essential Role in Human Carcinogenesis through Positive Regulation of Cyclin-Dependent Kinase. Cancer Prev. Res. 2011, 4, 2051–2061. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, C. Identification of a new class of PAX3-FKHR target promoters: A role of the Pax3 paired box DNA binding domain. Oncogene 2006, 26, 1595–1605. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Krieg, A.J. KDM4B: A Nail for Every Hammer? Genes 2019, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Ciarapica, R.; De Salvo, M.; Carcarino, E.; Bracaglia, G.; Adesso, L.; Leoncini, P.P.; Dall’Agnese, A.; Walters, Z.S.; Verginelli, F.; De Sio, L.; et al. The Polycomb group (PcG) protein EZH2 supports the survival of PAX3-FOXO1 alveolar rhabdomyosarcoma by repressing FBXO32 (Atrogin1/MAFbx). Oncogene 2013, 33, 4173–4184. [Google Scholar] [CrossRef] [Green Version]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.V.; Kala, M.P.; Rao, V.K.; Pignata, L.; Lim, H.J.; Suriyamurthy, S.; Chang, K.T.E.; Lee, V.K.; Guccione, E.; Taneja, R. Epigenetic Regulation of the PTEN–AKT–RAC1 Axis by G9a Is Critical for Tumor Growth in Alveolar Rhabdomyosarcoma. Cancer Res. 2019, 79, 2232–2243. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Altahan, A.M.; Hu, D.; Wang, Y.; Cheng, P.-H.; Morton, C.L.; Qu, C.; Nathwani, A.C.; Shohet, J.M.; Fotsis, T.; et al. The Role of Histone Demethylase KDM4B in Myc Signaling in Neuroblastoma. J. Natl. Cancer Inst. 2015, 107, djv080. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Milasta, S.; Ashutosh, M.; Altahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting Histone Demethylases in MYC-Driven Neuroblastomas with Ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [Green Version]
- Szakacs, G.; Annereau, J.P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.J.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef] [Green Version]
- von Manstein, V.; Yang, C.M.; Richter, D.; Delis, N.; Vafaizadeh, V.; Groner, B. Resistance of Cancer Cells to Targeted Therapies Through the Activation of Compensating Signaling Loops. Curr. Signal Transduct. Ther. 2013, 8, 193–202. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epidrug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Klose, R.J.; Yan, Q.; Tothova, Z.; Yamane, K.; Erdjument-Bromage, H.; Tempst, P.; Gilliland, D.G.; Zhang, Y.; Kaelin, W.G. The Retinoblastoma Binding Protein RBP2 Is an H3K4 Demethylase. Cell 2007, 128, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; A Tindell, C.; Wilson, C.; E Williamson, K.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Leurs, U.; Lohse, B.; Rand, K.D.; Ming, S.; Riise, E.S.; Cole, P.A.; Kristensen, J.L.; Clausen, R.P. Substrate- and Cofactor-independent Inhibition of Histone Demethylase KDM4C. ACS Chem. Biol. 2014, 9, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Manning, A.L.; Van Rechem, C.; Kim, J.; Ladd, B.; Cho, J.; Pineda, C.M.; Murphy, N.; Daniels, D.L.; Montagna, C.; et al. KDM4A Lysine Demethylase Induces Site-Specific Copy Gain and Rereplication of Regions Amplified in Tumors. Cell 2013, 154, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walters, Z.S.; Aladowicz, E.; Villarejo-Balcells, B.; Nugent, G.; Selfe, J.L.; Eve, P.; Blagg, J.; Rossanese, O.; Shipley, J. Role for the Histone Demethylase KDM4B in Rhabdomyosarcoma via CDK6 and CCNA2: Compensation by KDM4A and Apoptotic Response of Targeting Both KDM4B and KDM4A. Cancers 2021, 13, 1734. https://doi.org/10.3390/cancers13071734
Walters ZS, Aladowicz E, Villarejo-Balcells B, Nugent G, Selfe JL, Eve P, Blagg J, Rossanese O, Shipley J. Role for the Histone Demethylase KDM4B in Rhabdomyosarcoma via CDK6 and CCNA2: Compensation by KDM4A and Apoptotic Response of Targeting Both KDM4B and KDM4A. Cancers. 2021; 13(7):1734. https://doi.org/10.3390/cancers13071734
Chicago/Turabian StyleWalters, Zoë S., Ewa Aladowicz, Barbara Villarejo-Balcells, Gary Nugent, Joanna L. Selfe, Paul Eve, Julian Blagg, Olivia Rossanese, and Janet Shipley. 2021. "Role for the Histone Demethylase KDM4B in Rhabdomyosarcoma via CDK6 and CCNA2: Compensation by KDM4A and Apoptotic Response of Targeting Both KDM4B and KDM4A" Cancers 13, no. 7: 1734. https://doi.org/10.3390/cancers13071734