
RGL2 Drives the Metastatic Progression of Colorectal Cancer via Preventing the Protein Degradation of β-Catenin and KRAS

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
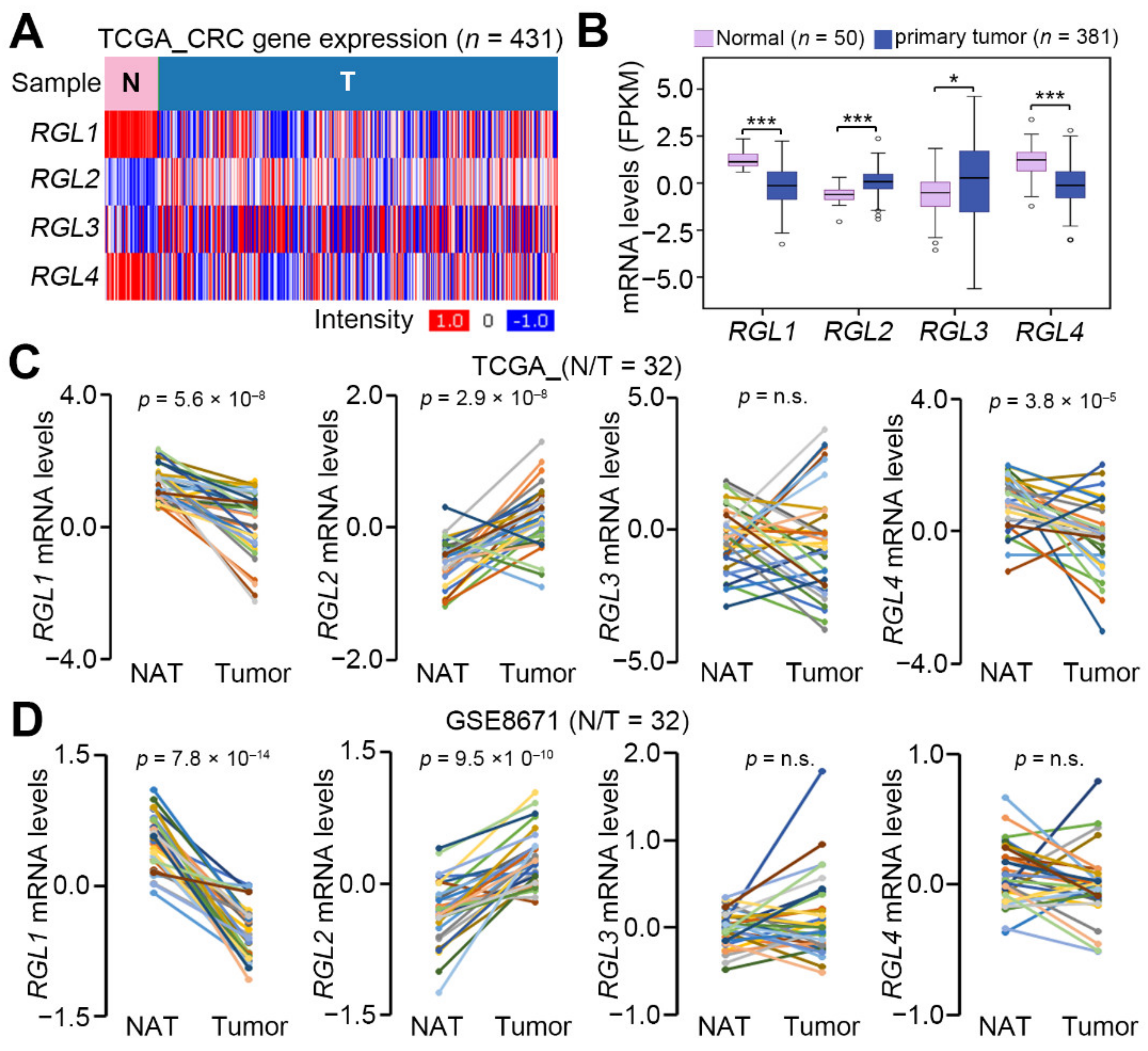
2.1. RGL2 Is Upregulated in Colorectal Cancer Compared to Normal Adjacent Tissues
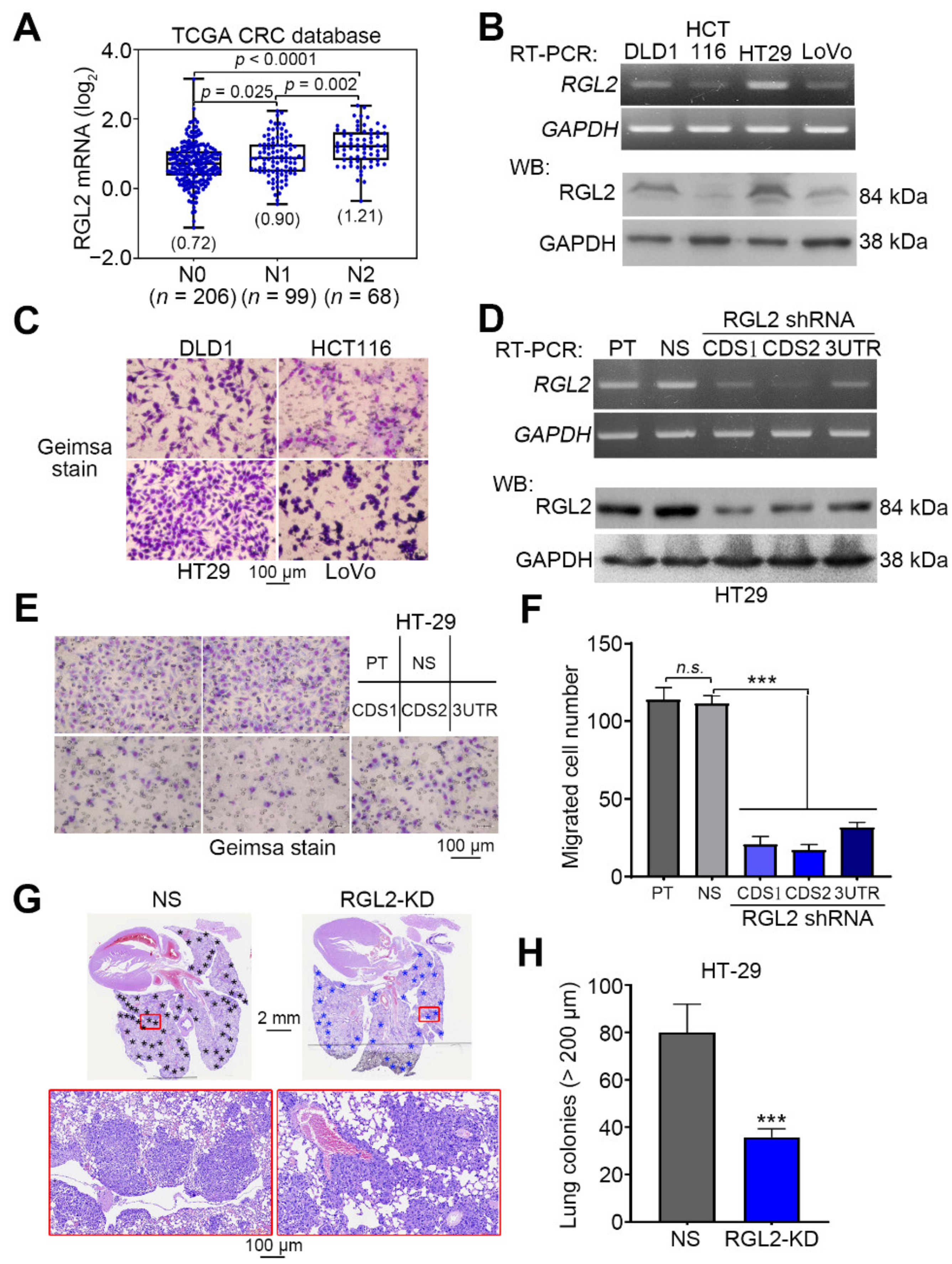
2.2. RGL2 Upregulation Correlates with a Poorer Prognosis in CRC Patients
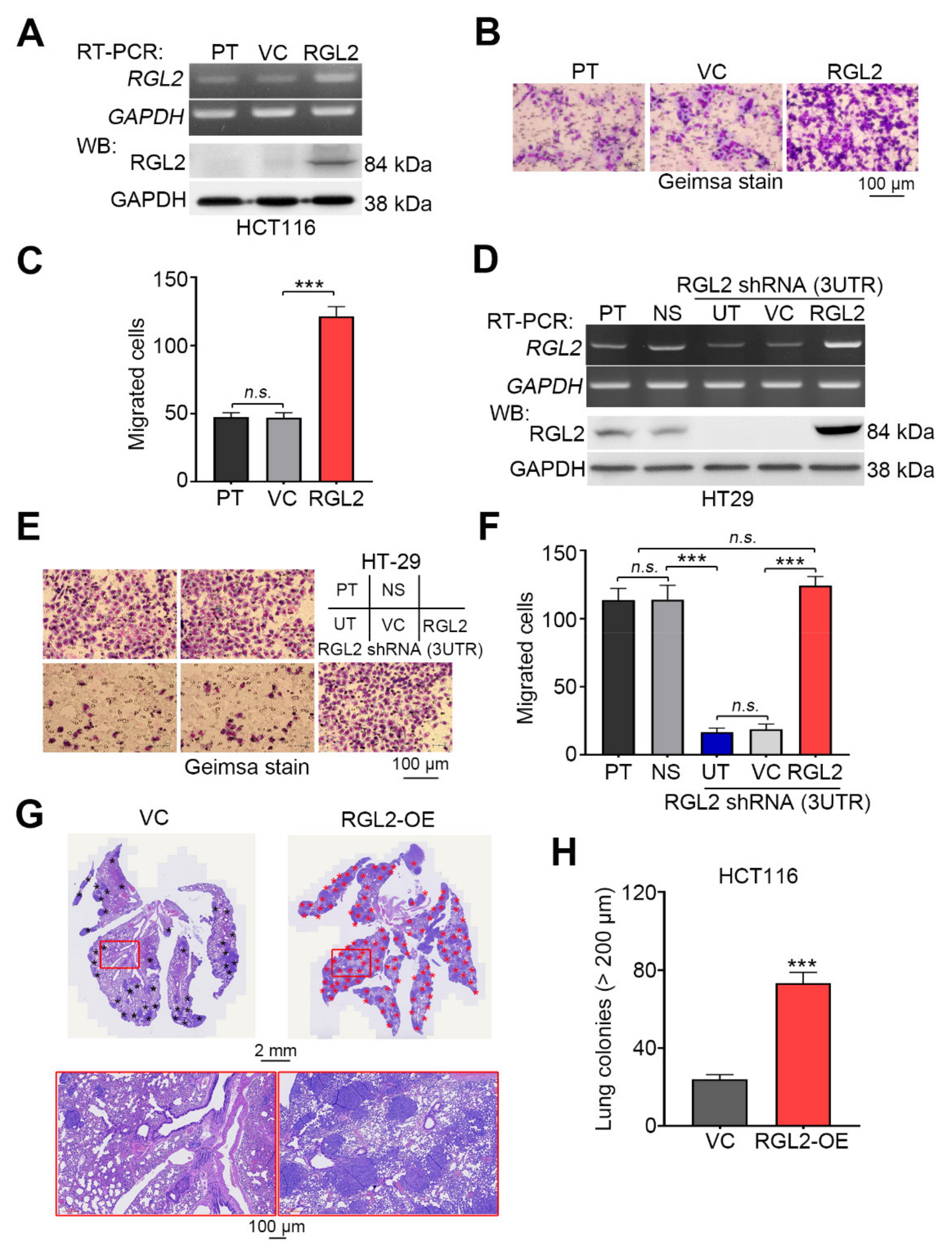
2.3. RGL2 Upregulation Promotes the Metastatic Progression of CRC Cells
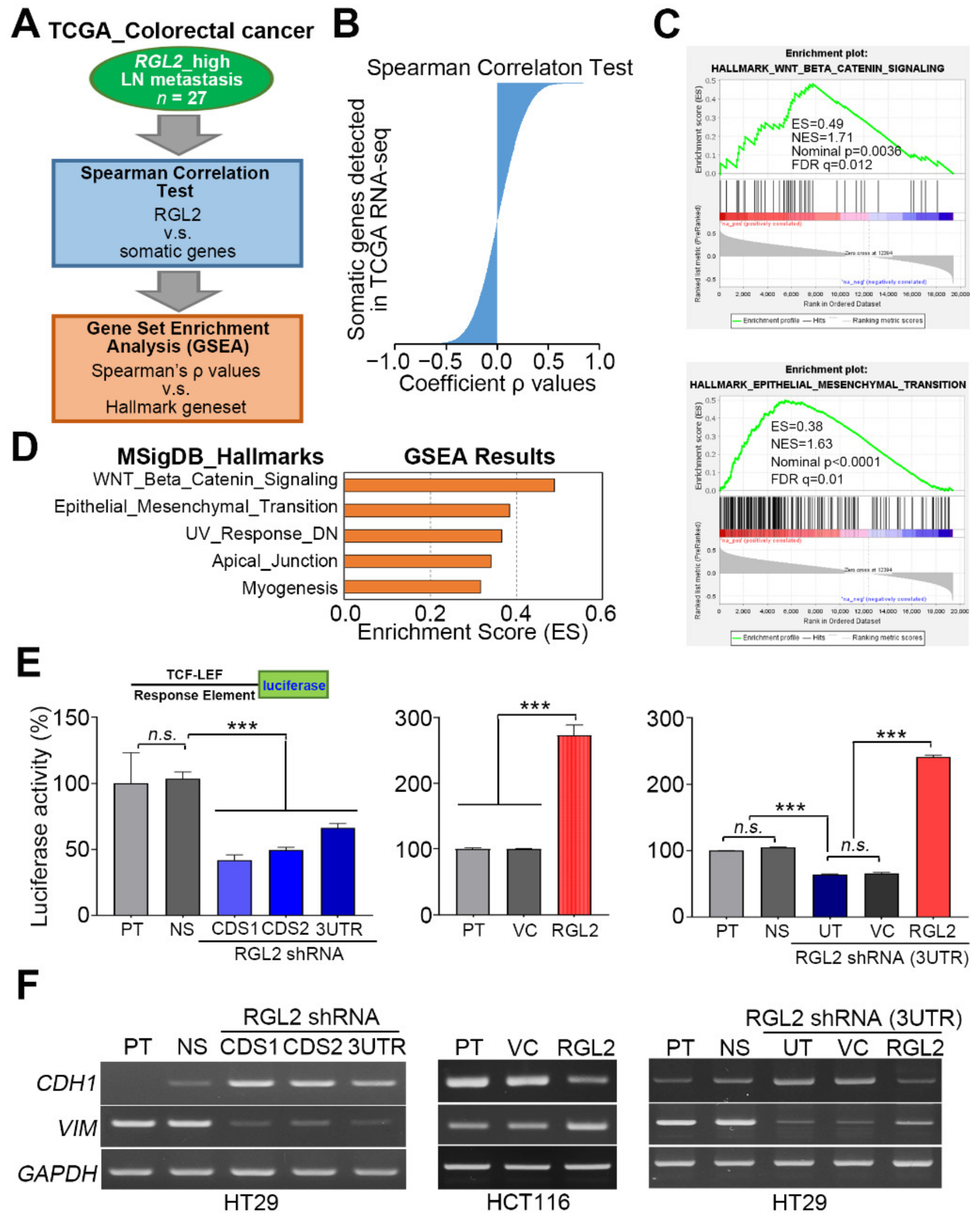
2.4. RGL2 Induces the Activation of the Wnt/β-Catenin Pathway and EMT Progression and Thereby Promotes CRC Metastasis
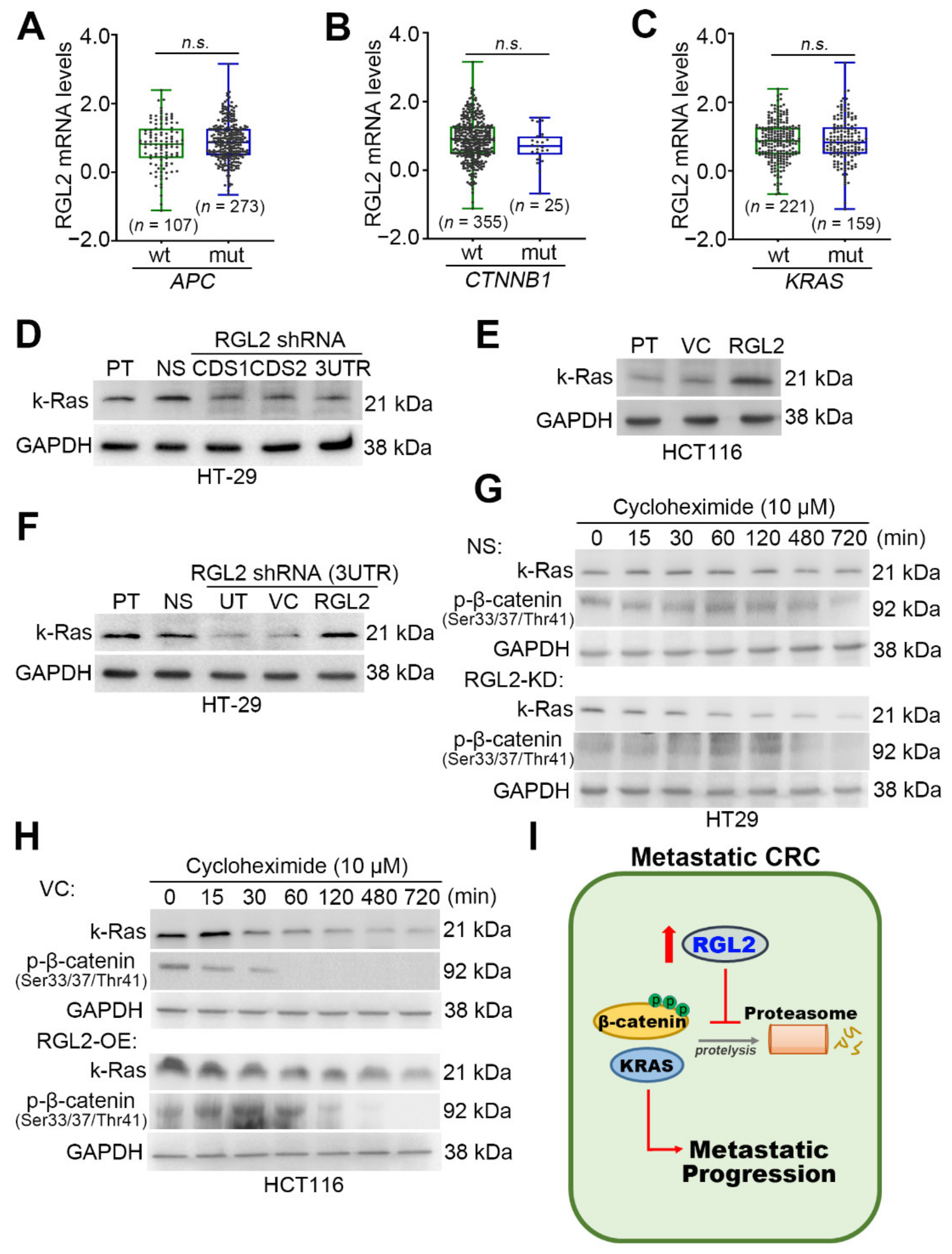
2.5. RGL2 Upregulation Enhances the Protein Stability of β-Catenin and KRAS in Metastatic CRC Cells
3. Discussion
4. Materials and Methods
4.1. Clinical and Molecular Data for CRC Patients
4.2. Cell Lines and Cell Culture Condition
4.3. MTT Assay
4.4. Plasmid Construction
4.5. Lentivirus-Driven shRNA Infection
4.6. Reverse Transcription PCR (RT-PCR)
4.7. Western Blotting Analysis
4.8. Promoter Reporter Assay
4.9. Migration Assay
4.10. Immunohistochemistry Staining
4.11. Animal Experiment
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.R.; Bergstralh, E.J.; Coffey, R.J.; Beart, R.W., Jr.; Melton, L.J., III. Predictors of survival after curative resection of carcinoma of the colon and rectum. Cancer 1987, 60, 2318–2324. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Chun, Y.J.; Kim, H.S.; Kim, J.H.; Lee, C.K.; Beom, S.H.; Shin, S.J.; Ahn, J.B. Clinical features and KRAS mutation in colorectal cancer with bone metastasis. Sci. Rep. 2020, 10, 21180–21188. [Google Scholar] [CrossRef]
- Chu, P.C.; Lin, P.C.; Wu, H.Y.; Lin, K.T.; Wu, C.; Bekaii-Saab, T.; Lin, Y.J.; Lee, C.T.; Lee, J.C.; Chen, C.S. Mutant KRAS promotes liver metastasis of colorectal cancer, in part, by upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR signaling pathway. Oncogene 2018, 37, 3440–3455. [Google Scholar] [CrossRef]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Rocken, C.; Lofton-Day, C.; Schulz, H.U.; Muller, O.; Kutzner, N.; Malfertheiner, P.; Ebert, M.P. Molecular analysis of APC promoter methylation and protein expression in colorectal cancer metastasis. Carcinogenesis 2005, 26, 37–43. [Google Scholar] [CrossRef]
- Jeong, W.J.; Ro, E.J.; Choi, K.Y. Interaction between Wnt/beta-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of beta-catenin and RAS by targeting the Wnt/beta-catenin pathway. NPJ. Precis. Oncol. 2018, 2, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, F.; Fields, S.; Schneider, C.; Martin, G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, Y.; Ren, S.; Li, X.; Yang, P.; Zhu, J.; Lin, L.; Wang, Z.; Jia, Y. Low expression of RGL4 is associated with a poor prognosis and immune infiltration in lung adenocarcinoma patients. Int. Immunopharmacol. 2020, 83, 106454–106465. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Martin, T.D.; Williams, F.; Yeh, J.J.; Campbell, S.L.; Der, C.J. Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J. Biol. Chem. 2010, 285, 34729–34740. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Jeong, W.J.; Cho, Y.H.; Cha, P.H.; Yoon, J.S.; Ro, E.J.; Choi, S.; Oh, J.M.; Heo, Y.; Kim, H.; et al. beta-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK3beta. EMBO Rep. 2018, 19, e46060–e46074. [Google Scholar] [CrossRef]
- Wu, G.; He, X. Threonine 41 in beta-catenin serves as a key phosphorylation relay residue in beta-catenin degradation. Biochemistry 2006, 45, 5319–5323. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Hamad, N.M.; Elconin, J.H.; Karnoub, A.E.; Bai, W.; Rich, J.N.; Abraham, R.T.; Der, C.J.; Counter, C.M. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002, 16, 2045–2057. [Google Scholar] [CrossRef] [Green Version]
- Ceriani, M.; Scandiuzzi, C.; Amigoni, L.; Tisi, R.; Berruti, G.; Martegani, E. Functional analysis of RalGPS2, a murine guanine nucleotide exchange factor for RalA GTPase. Exp. Cell Res. 2007, 313, 2293–2307. [Google Scholar] [CrossRef] [PubMed]
- Santos, O.; Parrini, M.C.; Camonis, J. RalGPS2 Is Essential for Survival and Cell Cycle Progression of Lung Cancer Cells Independently of Its Established Substrates Ral GTPases. PLoS ONE 2016, 11, e0154840–e0154859. [Google Scholar] [CrossRef]
- Lim, K.H.; O’Hayer, K.; Adam, S.J.; Kendall, S.D.; Campbell, P.M.; Der, C.J.; Counter, C.M. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 2006, 16, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Zago, G.; Veith, I.; Singh, M.K.; Fuhrmann, L.; De, B.S.; Remorino, A.; Takaoka, S.; Palmeri, M.; Berger, F.; Brandon, N.; et al. RalB directly triggers invasion downstream Ras by mobilizing the Wave complex. Elife 2018, 7, e40474–e40496. [Google Scholar] [CrossRef]
- Fernandez, R.M.; Ruiz-Miro, M.; Dolcet, X.; Aldea, M.; Gari, E. Cyclin D1 interacts and collaborates with Ral GTPases enhancing cell detachment and motility. Oncogene 2011, 30, 1936–1946. [Google Scholar] [CrossRef]
- Cheriyamundath, S.; Ben-Ze’ev, A. Wnt/beta-Catenin Target Genes in Colon Cancer Metastasis: The Special Case of L1CAM. Cancers 2020, 12, 3444. [Google Scholar] [CrossRef]
- Jeong, W.J.; Yoon, J.; Park, J.C.; Lee, S.H.; Lee, S.H.; Kaduwal, S.; Kim, H.; Yoon, J.B.; Choi, K.Y. Ras stabilization through aberrant activation of Wnt/beta-catenin signaling promotes intestinal tumorigenesis. Sci. Signal. 2012, 5, ra30–ra42. [Google Scholar] [CrossRef]
- Moon, B.S.; Jeong, W.J.; Park, J.; Kim, T.I.; Min, D.S.; Choi, K.Y. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/beta-catenin signaling. J. Natl. Cancer Inst. 2014, 106, djt373–djt382. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.-S.; Yuan, L.-T.; Kuei, C.-H.; Lin, H.-Y.; Chen, Y.-L.; Chiu, H.-W.; Lin, Y.-F. RGL2 Drives the Metastatic Progression of Colorectal Cancer via Preventing the Protein Degradation of β-Catenin and KRAS. Cancers 2021, 13, 1763. https://doi.org/10.3390/cancers13081763
Sun M-S, Yuan L-T, Kuei C-H, Lin H-Y, Chen Y-L, Chiu H-W, Lin Y-F. RGL2 Drives the Metastatic Progression of Colorectal Cancer via Preventing the Protein Degradation of β-Catenin and KRAS. Cancers. 2021; 13(8):1763. https://doi.org/10.3390/cancers13081763
Chicago/Turabian StyleSun, Meng-Shun, Lan-Ting Yuan, Chia-Hao Kuei, Hui-Yu Lin, Yen-Lin Chen, Hui-Wen Chiu, and Yuan-Feng Lin. 2021. "RGL2 Drives the Metastatic Progression of Colorectal Cancer via Preventing the Protein Degradation of β-Catenin and KRAS" Cancers 13, no. 8: 1763. https://doi.org/10.3390/cancers13081763