
SIO: A Spatioimageomics Pipeline to Identify Prognostic Biomarkers Associated with the Ovarian Tumor Microenvironment

 , ,
, ,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Preparation and Staining
2.3. Imaging Mass Cytometry
2.4. Microdissection and Microarray Analysis of Tissue Samples
2.5. Data Preprocessing and Cell Segmentation
2.6. Analysis Workflow
2.7. Clustering Analysis
2.8. Cell Density and Nearest-Neighbor Interactions in Tumor-Enriched Regions
2.9. Survival Prediction
2.10. Correlation of Cell Density with Gene Expression
2.11. Kaplan-Meier Analysis
2.12. Data and Code Availability
3. Results
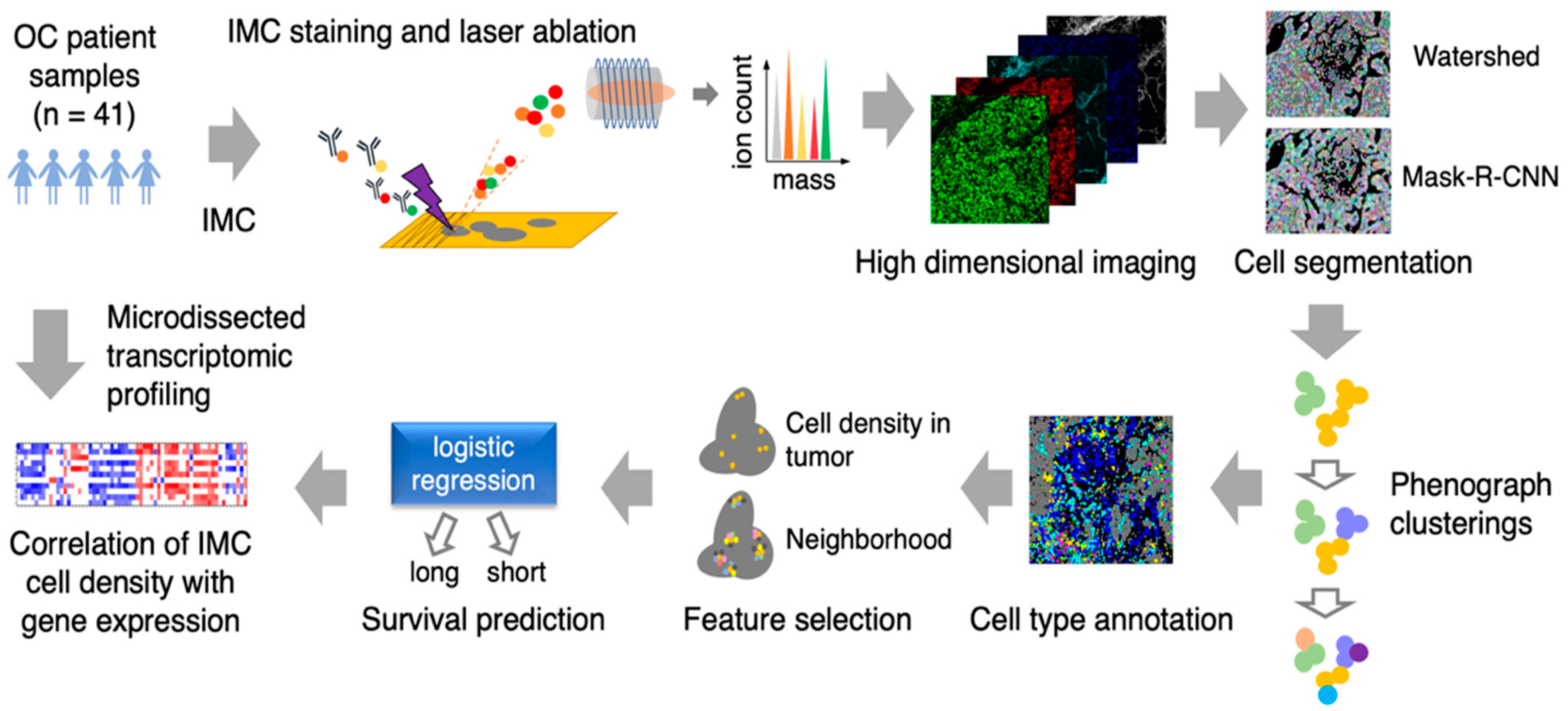
3.1. Image Analysis Pipeline
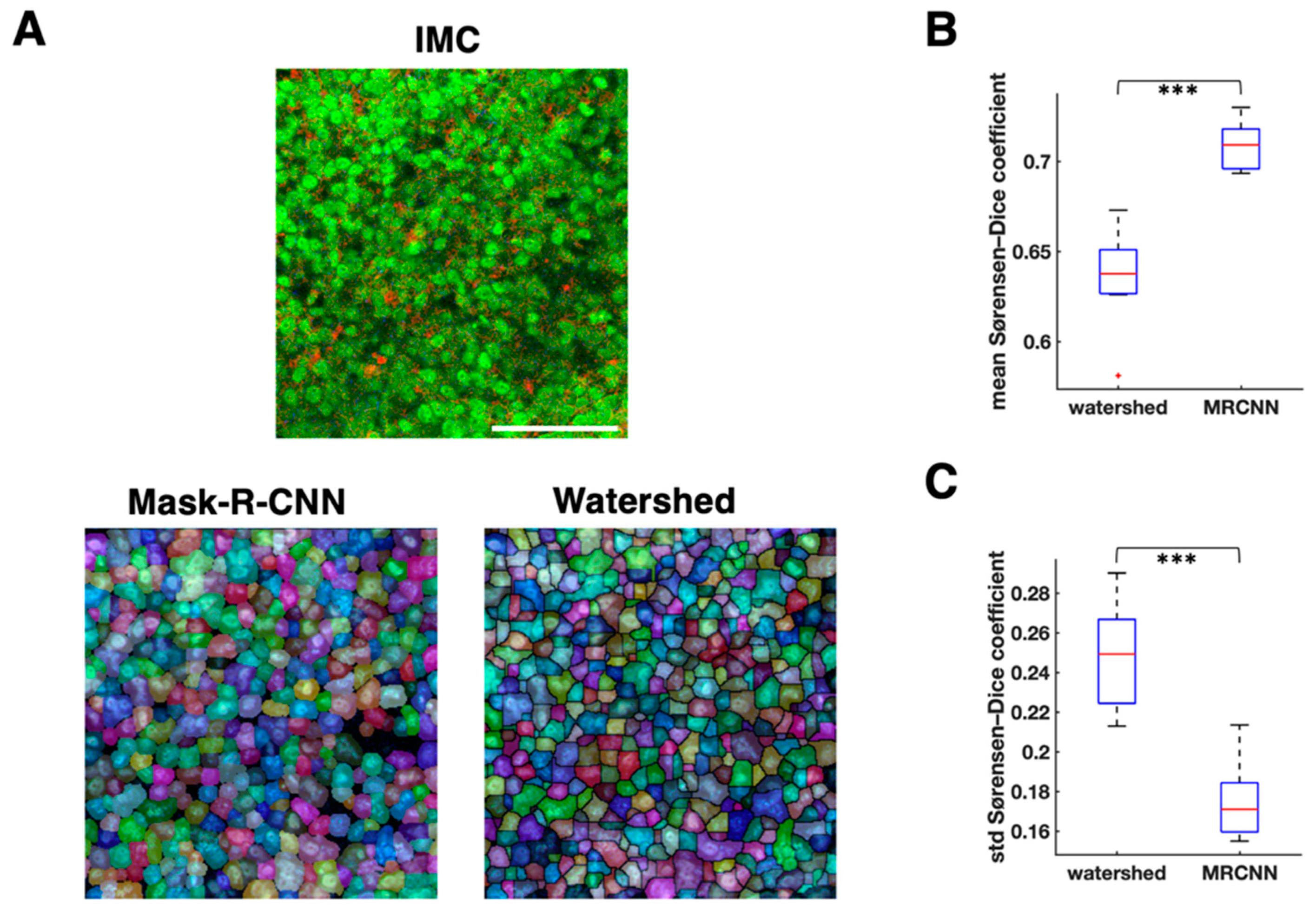
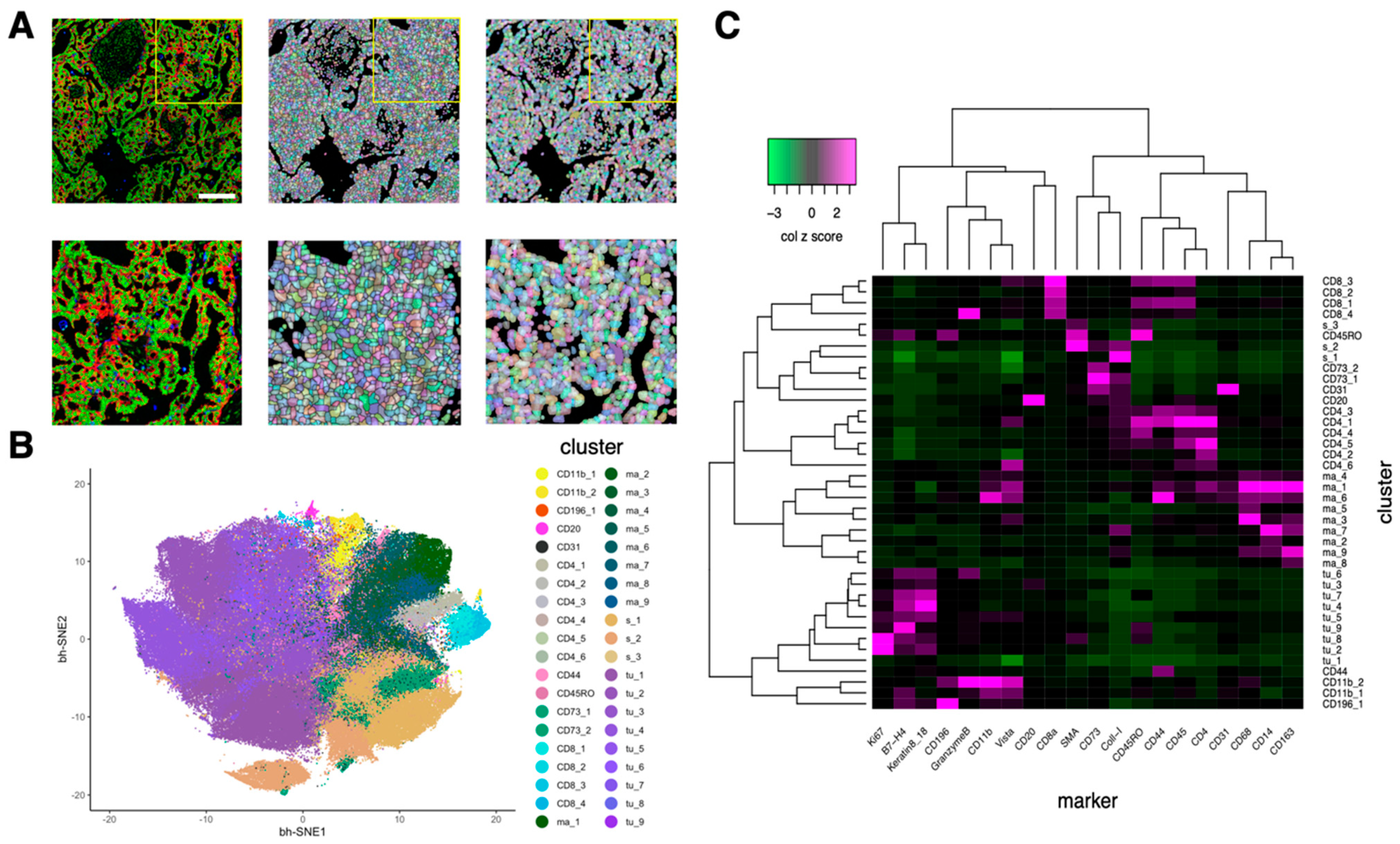
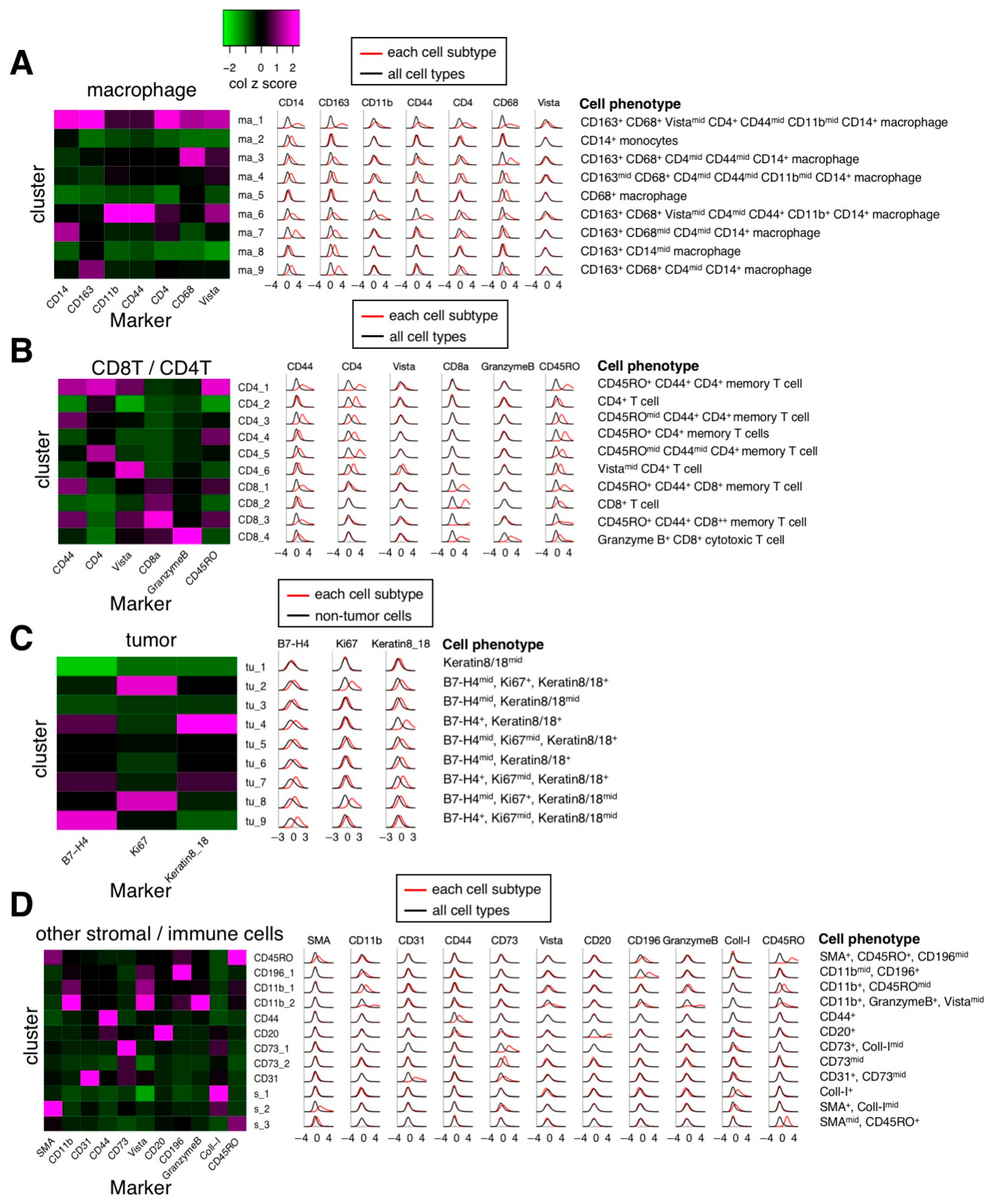
3.2. Cell Segmentation and Annotation by Deep Learning-Based IMC Data Analysis
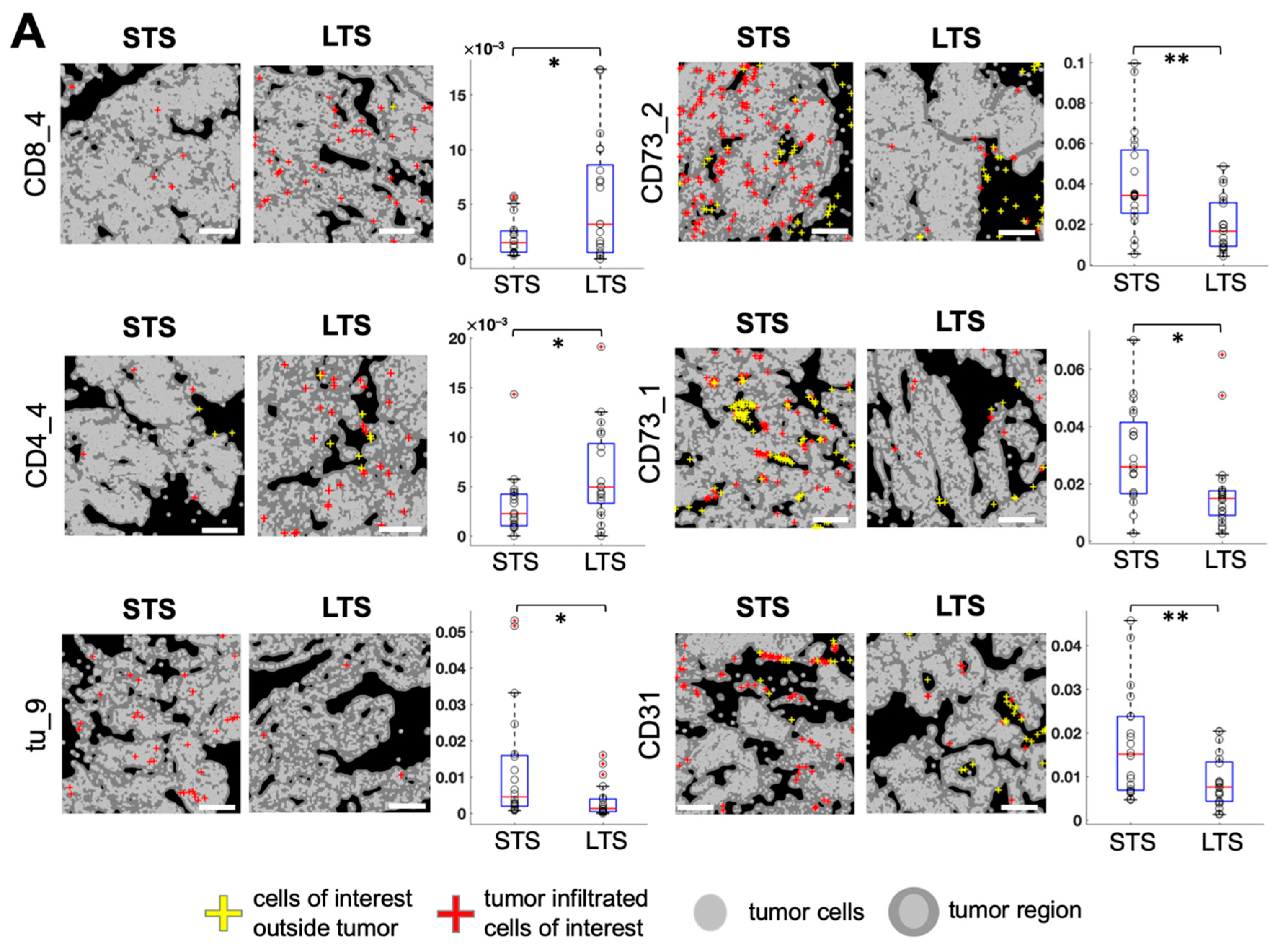
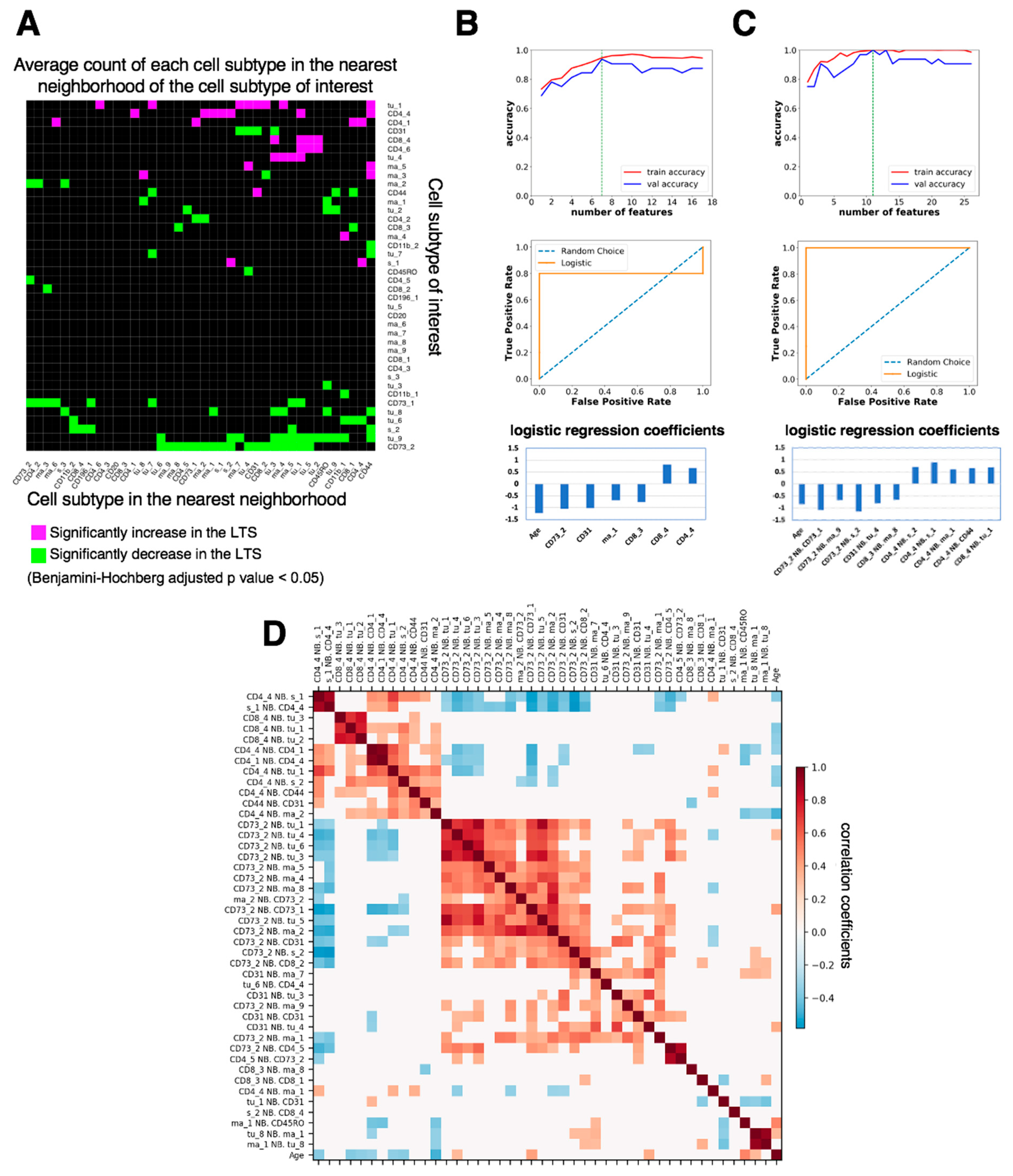
3.3. Spatially Resolved Cell Density and Nearest-Neighbor Cell-Cell Interactions Analyses of the Ovarian Tumor Microenvironment
3.4. Feature Selection for Overall Survival Prediction by Logistic Regression
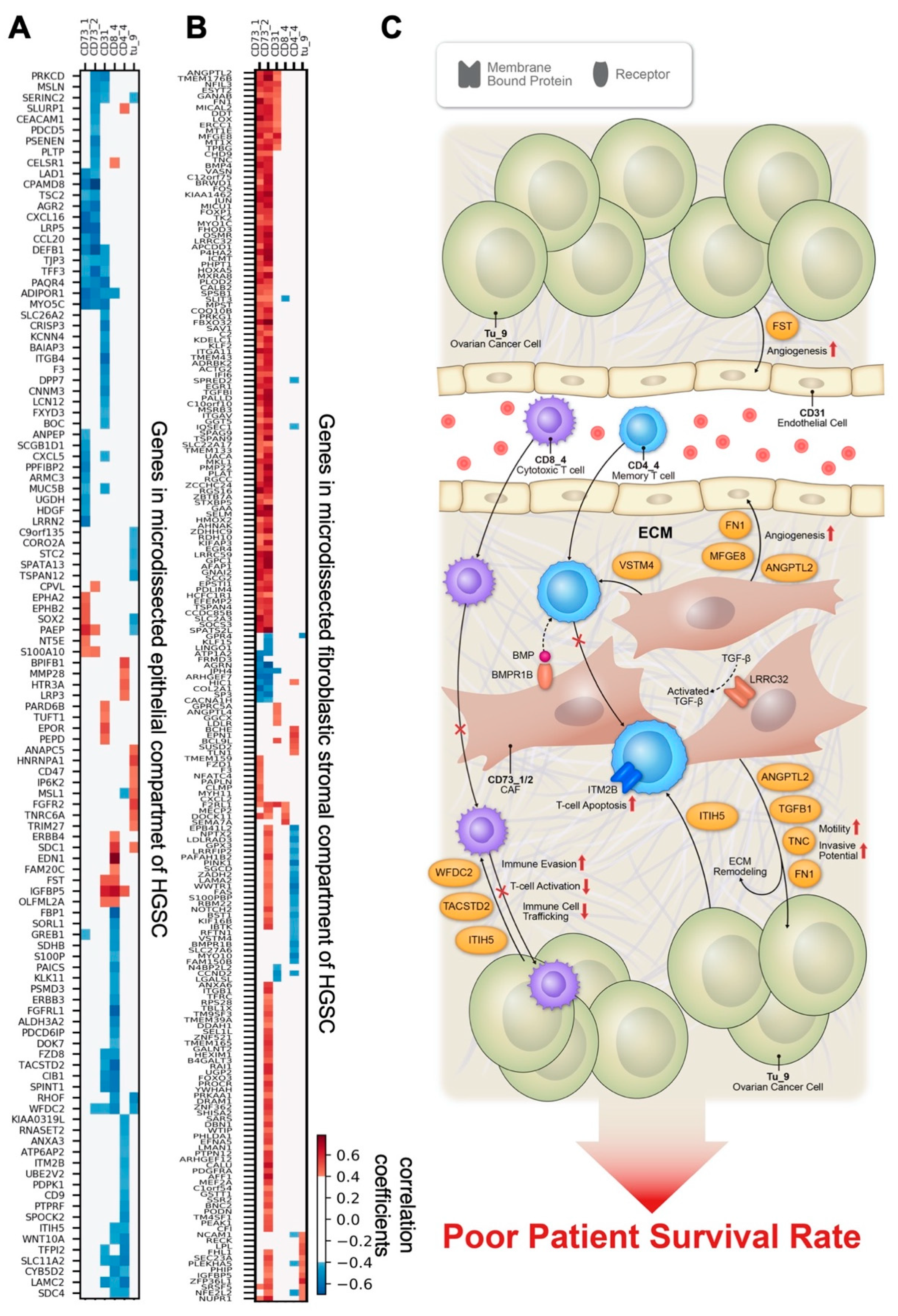
3.5. Correlations between Cell Subtype Density and Transcriptomic Profiles from Microdissected Fibroblastic Stromal and Epithelial Compartments of HGSC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef]
- Sato, S.; Itamochi, H. Neoadjuvant chemotherapy in advanced ovarian cancer: Latest results and place in therapy. Ther. Adv. Med. Oncol. 2014, 6, 293–304. [Google Scholar] [CrossRef]
- Ushijima, K. Treatment for Recurrent Ovarian Cancer—At First Relapse. J. Oncol. 2009, 2010, 1–7. [Google Scholar] [CrossRef]
- Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.-H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2016, 8, 3933–3945. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer 2019, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef]
- Chang, A.Y.; Bhattacharya, N.; Mu, J.; Setiadi, A.F.; Carcamo-Cavazos, V.; Lee, G.H.; Simons, D.L.; Yadegarynia, S.; Hemati, K.; Kapelner, A.; et al. Spatial organization of dendritic cells within tumor draining lymph nodes impacts clinical outcome in breast cancer pa-tients. J. Transl. Med. 2013, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Feichtenbeiner, A.; Haas, M.; Büttner, M.; Grabenbauer, G.G.; Fietkau, R.; Distel, L.V. Critical role of spatial interaction between CD8+ and Foxp3+ cells in human gastric cancer: The distance matters. Cancer Immunol. Immunother. 2013, 63, 111–119. [Google Scholar] [CrossRef]
- Setiadi, A.F.; Ray, N.C.; Kohrt, H.E.; Kapelner, A.; Carcamo-Cavazos, V.; Levic, E.B.; Yadegarynia, S.; Van Der Loos, C.M.; Schwartz, E.J.; Holmes, S.; et al. Quantitative, architectural analysis of immune cell subsets in tumor-draining lymph nodes from breast cancer patients and healthy lymph nodes. PLoS ONE 2010, 5, e12420. [Google Scholar] [CrossRef]
- Yuan, Y.; Failmezger, H.; Rueda, O.M.; Ali, H.R.; Gräf, S.; Chin, S.F.; Schwarz, R.F.; Curtis, C.; Dunning, M.J.; Bardwell, H.; et al. Quantitative image analysis of cellular heterogeneity in breast tumors comple-ments genomic profiling. Sci. Transl. Med. 2012, 4, 157ra143. [Google Scholar] [CrossRef]
- Schapiro, D.; Jackson, H.W.; Raghuraman, S.; Fischer, J.R.; Zanotelli, V.R.; Schulz, D.; Giesen, C.; Catena, R.; Varga, Z.; Bodenmiller, B. miCAT: A toolbox for analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods 2017, 14, 873–876. [Google Scholar] [CrossRef]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell pathology landscape of breast cancer. Nat. Cell Biol. 2020, 578, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; El-ad, D.A.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef]
- He, K.; Gkioxari, G.; Dollár, P.; Girshick, R. Mask R-CNN. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 42, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Maire, M.; Belongie, S.; Hays, J.; Perona, P.; Ramanan, D.; Dollár, P.; Zitnick, C.L. Microsoft COCO: Common objects in context. In Computer Vision—ECCV 2014. ECCV 2014. Lecture Notes in Computer Science; Springer: Cham, Swizerland, 2014; pp. 740–755. [Google Scholar]
- Johnson, J.W. Adapting Mask-RCNN for Automatic Nucleus Segmentation. arXiv 2020, arXiv:180500500. [Google Scholar]
- Shih, A.J.; Menzin, A.; Whyte, J.; Lovecchio, J.; Liew, A.; Khalili, H.; Bhuiya, T.; Gregersen, P.K.; Lee, A.T. Identification of grade and origin specific cell populations in serous epithelial ovarian cancer by single cell RNA-seq. PLoS ONE 2018, 13, e0206785. [Google Scholar]
- Yeung, T.-L.; Sheng, J.; Leung, C.S.; Li, F.; Kim, J.; Ho, S.Y.; Matzuk, M.M.; Lu, K.H.; Wong, S.T.C.; Mok, S.C. Systematic Identification of Druggable Epithelial–Stromal Crosstalk Signaling Networks in Ovarian Cancer. J. Natl. Cancer Inst. 2019, 111, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Crowell, H.L.; Zanotelli, V.R.; Engler, S.; Robinson, M.D.; Bodenmiller, B. Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst. 2018, 6, 612–620.e5. [Google Scholar] [CrossRef]
- Novo, D.; Grégori, G.; Rajwa, B. Generalized unmixing model for multispectral flow cytometry utilizing nonsquare compensation matrices. Cytom. Part A 2013, 83, 508–520. [Google Scholar] [CrossRef] [PubMed]
- van der Maaten, L. Barnes-Hut-SNE. arXiv 2013, arXiv:13013342. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Győrffy, B.; Lánczky, A.; Szállási, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fushida, S.; Yamamoto, Y.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Miyashita, T.; Tajima, H.; Ninomiya, I.; Munesue, S.; et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer 2016, 19, 1052–1065. [Google Scholar] [CrossRef]
- Koelzer, V.H.; Canonica, K.; Dawson, H.; Sokol, L.; Karamitopoulou-Diamantis, E.; Lugli, A.; Zlobec, I. Phenotyping of tumor-associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology 2015, 5, e1106677. [Google Scholar] [CrossRef]
- Minami, K.; Hiwatashi, K.; Ueno, S.; Sakoda, M.; Iino, S.; Okumura, H.; Hashiguchi, M.; Kawasaki, Y.; Kurahara, H.; Mataki, Y.; et al. Prognostic significance of CD68, CD163 and Folate receptor-β positive macrophages in hepatocellular carcinoma. Exp. Ther. Med. 2018, 15, 4465–4476. [Google Scholar] [CrossRef]
- Wei, F.; Wu, Y.; Tang, L.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Liao, Q.; et al. BPIFB1 (LPLUNC1) inhibits migration and invasion of nasopharyngeal carcinoma by interacting with VTN and VIM. Br. J. Cancer 2018, 118, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Ikeyama, S.; Koyama, M.; Yamaoko, M.; Sasada, R.; Miyake, M. Suppression of cell motility and metastasis by transfection with human motility-related protein (MRP-1/CD9) DNA. J. Exp. Med. 1993, 177, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.R.; Jo, K.; Lee, Y.; Sung, B.J.; Park, Y.W.; Lee, J.H. Upregulation of CD9 in ovarian cancer is related to the induction of TNF-α gene expression and constitutive NF-κB activation. Carcinogenesis 2012, 33, 77–83. [Google Scholar] [CrossRef]
- Veeck, J.; Chorovicer, M.; Naami, A.; Breuer, E.; Zafrakas, M.; Bektas, N.; Dürst, M.; Kristiansen, G.; Wild, P.J.; Hartmann, A.; et al. The extracellular matrix protein ITIH5 is a novel prognostic marker in invasive node-negative breast cancer and its aberrant expression is caused by promoter hypermethylation. Oncogene 2007, 27, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Ridge, R.J.; Sloane, N.H. Partial N-terminal amino acid sequence of the anti-neoplastic urinary protein (ANUP) and the anti-tumour effect of the N-terminal nonapeptide of the unique cytokine present in human granulocytes. Cytokine 1996, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, Y.; Yoshikawa, K.; Fukuda, H.; Fujii, Y.X.; Misawa, H.; Kawashima, K. Immune system expression of SLURP-1 and SLURP-2, two endogenous nicotinic acetylcholine receptor ligands. Life Sci. 2007, 80, 2365–2368. [Google Scholar] [CrossRef]
- Feng, Y.; Gao, Y.; Yu, J.; Jiang, G.; Zhang, X.; Lin, X.; Han, Q.; Rong, X.; Xu, H.; Li, Q.; et al. CCDC85B promotes non-small cell lung cancer cell proliferation and invasion. Mol. Carcinog. 2019, 58, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Hulin, J.-A.; Tommasi, S.; Elliot, D.; Mangoni, A.A. Small molecule inhibition of DDAH1 significantly attenuates triple negative breast cancer cell vasculogenic mimicry in vitro. Biomed. Pharmacother. 2019, 111, 602–612. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Z.; Fang, S.; Fang, R.; Liu, X.; Zhao, Y.; Li, X.; Huang, L.; Zhang, J. Fibulin-4 is associated with tumor progression and a poor prognosis in ovarian carcinomas. BMC Cancer 2015, 15, 91. [Google Scholar] [CrossRef]
- Ungefroren, H.; Witte, D.; Rauch, B.H.; Settmacher, U.; Lehnert, H.; Gieseler, F.; Kaufmann, R. Proteinase-Activated Receptor 2 May Drive Cancer Progression by Facilitating TGF-β Signaling. Int. J. Mol. Sci. 2017, 18, 2494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zou, W. Inhibition of integrin β1 decreases the malignancy of ovarian cancer cells and potentiates anticancer therapy via the FAK/STAT1 signaling pathway. Mol. Med. Rep. 2015, 12, 7869–7876. [Google Scholar] [CrossRef]
- Ji, F.; Wang, Y.; Qiu, L.; Li, S.; Zhu, J.; Liang, Z.; Wan, Y.; Di, W. Hypoxia inducible factor 1α-mediated LOX expression correlates with migration and invasion in epithelial ovarian cancer. Int. J. Oncol. 2013, 42, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Uchiyama, A.; Uehara, A.; Perera, B.; Ogino, S.; Yokoyama, Y.; Takeuchi, Y.; Udey, M.C.; Ishikawa, O.; Motegi, S.-I. MFG-E8 Drives Melanoma Growth by Stimulating Mesenchymal Stromal Cell–Induced Angiogenesis and M2 Polarization of Tumor-Associated Macrophages. Cancer Res. 2016, 76, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, A.; Yamada, K.; Ogino, S.; Yokoyama, Y.; Takeuchi, Y.; Udey, M.C.; Ishikawa, O.; Motegi, S.-I. MFG-E8 Regulates Angiogenesis in Cutaneous Wound Healing. Am. J. Pathol. 2014, 184, 1981–1990. [Google Scholar] [CrossRef]
- Zhu, L.-Y.; Zhang, W.-M.; Yang, X.-M.; Cui, L.; Li, J.; Zhang, Y.-L.; Wang, Y.-H.; Ao, J.-P.; Ma, M.-Z.; Lu, H.; et al. Silencing of MICAL-L2 suppresses malignancy of ovarian cancer by inducing mesenchymal–epithelial transition. Cancer Lett. 2015, 363, 71–82. [Google Scholar] [CrossRef]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef]
- Zecchini, S.; Bombardelli, L.; Decio, A.; Bianchi, M.; Mazzarol, G.; Sanguineti, F.; Aletti, G.; Maddaluno, L.; Berezin, V.; Bock, E.; et al. The adhesion molecule NCAM promotes ovarian cancer progression via FGFR signalling. EMBO Mol. Med. 2011, 3, 480–494. [Google Scholar] [CrossRef]
- Xu, C.; Tian, G.; Jiang, C.; Xue, H.; Kuerbanjiang, M.; Sun, L.; Gu, L.; Zhou, H.; Liu, Y.; Zhang, Z.; et al. NPTX2 promotes colorectal cancer growth and liver metastasis by the activation of the canonical Wnt/β-catenin pathway via FZD6. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Teliga-Czajkowska, J.; Sienko, J.; Jalinik, K.; Smolarczyk, R.; Czajkowski, K. Prognostic value of tissue plasminogen activator (tPA) in patients with epithelial ovarian cancer undergoing chemotherapy. Ginekol. Polska 2016, 90, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ancey, P.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Kim, H.J.; Kim, M.K.; Bae, M.K.; Sung, H.Y.; Ahn, J.H.; Kim, Y.H.; Kim, S.C.; Ju, W. SPSB1 enhances ovarian cancer cell survival by destabilizing p21. Biochem. Biophys. Res. Commun. 2019, 510, 364–369. [Google Scholar] [CrossRef]
- Liang, W.; Guo, B.; Ye, J.; Liu, H.; Deng, W.; Lin, C.; Zhong, X.; Wang, L. Vasorin stimulates malignant progression and angiogenesis in glioma. Cancer Sci. 2019, 110, 2558–2572. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, P.; Mucha, O.; Józkowicz, A.; Dulak, J.; Łoboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Współczesna Onkol. 2018, 2018, 23–32. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, J.; Fu, B.; Dai, J.; Zhao, Y.; Lai, L. Isoprenylcysteine carboxylmethyltransferase regulates ovarian cancer cell response to chemotherapy and Ras activation. Biochem. Biophys. Res. Commun. 2018, 501, 556–562. [Google Scholar] [CrossRef]
- Qi, Y.; Qi, W.; Liu, S.; Sun, L.; Ding, A.; Yu, G.; Li, H.; Wang, Y.; Qiu, W.; Lv, J. TSPAN9 suppresses the chemosensitivity of gastric cancer to 5-fluorouracil by promoting autophagy. Cancer Cell Int. 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Aoi, J.; Endo, M.; Kadomatsu, T.; Miyata, K.; Nakano, M.; Horiguchi, H.; Ogata, A.; Odagiri, H.; Yano, M.; Araki, K.; et al. Angiopoietin-like Protein 2 Is an Important Facilitator of Inflammatory Carcinogenesis and Metastasis. Cancer Res. 2011, 71, 7502–7512. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef]
- Cao, R.; Chen, J.; Zhang, X.; Zhai, Y.; Qing, X.; Xing, W.; Zhang, L.; Malik, Y.S.; Yu, H.; Zhu, X. Elevated expression of myosin X in tumours contributes to breast cancer aggressiveness and metastasis. Br. J. Cancer 2014, 111, 539–550. [Google Scholar] [CrossRef]
- Zhang, X.; Fang, Q.; Ma, Y.; Zou, S.; Liu, Q.; Wang, H. Protease activated receptor 2 mediates tryptase-induced cell migration through MYO10 in colorectal cancer. Am. J. Cancer Res 2019, 9, 1995–2006. [Google Scholar]
- Wang, J.; Manick, B.; Renelt, M.; Suin, J.; Hansen, L.; Person, A.; Kalabokis, V.; Wu, G. VSTM4 is a novel negative regulator of T cell activation. J. Immunol. 2019, 202, 124.4. [Google Scholar]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.-L.; Leung, C.S.; Wong, K.-K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β Modulates Ovarian Cancer Invasion by Upregulating CAF-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Didem, T.; Faruk, T.; Senem, K.; Derya, D.; Murat, S.; Murat, G.; Oznur, K. Clinical significance of serum tenascin-c levels in epithelial ovarian cancer. Tumor Biol. 2014, 35, 6777–6782. [Google Scholar] [CrossRef]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of FAK–PI3K/Akt pathway. Cell Biol. Int. 2014, 38, 85–91. [Google Scholar] [CrossRef]
- Raymond, A.; Ensslin, M.A.; Shur, B.D. SED1/MFG-E8: A Bi-Motif protein that orchestrates diverse cellular interactions. J. Cell. Biochem. 2009, 106, 957–966. [Google Scholar] [CrossRef]
- Hwang, W.-T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Powell, D.J. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Abiko, K.; Matsumura, N.; Baba, T.; Yoshioka, Y.; Kosaka, K.; Konishi, I. The comprehensive assessment of local immune status of ovarian cancer by the clustering of multiple immune factors. Clin. Immunol. 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig. 2011, 121, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.; Haibe-Kains, B.; Karn, T.; et al. CD73 Is Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer. Cancer Res. 2015, 75, 4494–4503. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.-S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Liang, L.; Jiang, Y.; Chen, J.-S.; Niu, N.; Piao, J.; Ning, J.; Zu, Y.; Zhang, J.; Liu, J. B7-H4 expression in ovarian serous carcinoma: A study of 306 cases. Hum. Pathol. 2016, 57, 1–6. [Google Scholar] [CrossRef]
- Rask, L.; Høgdall, C.K.; Kjaer, S.K.; Christensen, L.; Jensen, A.; Blaakaer, J.; Christensen, I.J.; Høgdall, E.V. Association of CD31 and p53 With Survival of Ovarian Cancer Patients. Anticancer Res. 2019, 39, 567–576. [Google Scholar] [CrossRef]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Jackson, H.W.; Zanotelli, V.R.; Danenberg, E.; Fischer, J.R.; Bardwell, H.; Provenzano, E.; Rueda, O.M.; Chin, S.F.; Aparicio, S.; et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 2020, 1, 163–175. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef]
- Moore, R.G.; Hill, E.K.; Horan, T.; Yano, N.; Kim, K.; MacLaughlan, S.; Lambert-Messerlian, G.; Tseng, Y.; Padbury, J.F.; Miller, M.C.; et al. HE4 (WFDC2) gene overexpression promotes ovarian tumor growth. Sci. Rep. 2014, 4, 3574. [Google Scholar] [CrossRef]
- James, N.E.; Oliver, M.T.; Ribeiro, J.R.; Cantillo, E.; Rowswell-Turner, R.B.; Kim, K.K.; Chichester, C.O., III; DiSilvestro, P.A.; Moore, R.G.; Singh, R.K.; et al. Human Epididymis Secretory Protein 4 (HE4) Compromises Cytotoxic Mononuclear Cells via Inducing Dual Specificity Phosphatase 6. Front. Pharmacol. 2019, 10, 216. [Google Scholar] [CrossRef]
- Fleischer, A.; Ayllón, V.; Dumoutier, L.; Renauld, J.-C.; Rebollo, A. Proapoptotic activity of ITM2Bs, a BH3-only protein induced upon IL-2-deprivation which interacts with Bcl-2. Oncogene 2002, 21, 3181–3189. [Google Scholar] [CrossRef]
- Steitz, A.M.; Steffes, A.; Finkernagel, F.; Unger, A.; Sommerfeld, L.; Jansen, J.M.; Wagner, U.; Graumann, J.; Muller, R.; Reinartz, S. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, J.; Dong, X.; Shi, M.; Lu, C.; Springer, T.A. GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 2012, 23, 1129–1139. [Google Scholar] [CrossRef]
- Katsuta, E.; Maawy, A.A.; Yan, L.; Takabe, K. High expression of bone morphogenetic protein (BMP) 6 and BMP7 are associated with higher immune cell infiltration and better survival in estrogen receptor-positive breast cancer. Oncol. Rep. 2019, 42, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Ferri-Borgogno, S.; Sheng, J.; Yeung, T.-L.; Burks, J.K.; Cappello, P.; Jazaeri, A.A.; Kim, J.-H.; Han, G.H.; Birrer, M.J.; et al. SIO: A Spatioimageomics Pipeline to Identify Prognostic Biomarkers Associated with the Ovarian Tumor Microenvironment. Cancers 2021, 13, 1777. https://doi.org/10.3390/cancers13081777
Zhu Y, Ferri-Borgogno S, Sheng J, Yeung T-L, Burks JK, Cappello P, Jazaeri AA, Kim J-H, Han GH, Birrer MJ, et al. SIO: A Spatioimageomics Pipeline to Identify Prognostic Biomarkers Associated with the Ovarian Tumor Microenvironment. Cancers. 2021; 13(8):1777. https://doi.org/10.3390/cancers13081777
Chicago/Turabian StyleZhu, Ying, Sammy Ferri-Borgogno, Jianting Sheng, Tsz-Lun Yeung, Jared K. Burks, Paola Cappello, Amir A. Jazaeri, Jae-Hoon Kim, Gwan Hee Han, Michael J. Birrer, and et al. 2021. "SIO: A Spatioimageomics Pipeline to Identify Prognostic Biomarkers Associated with the Ovarian Tumor Microenvironment" Cancers 13, no. 8: 1777. https://doi.org/10.3390/cancers13081777
APA StyleZhu, Y., Ferri-Borgogno, S., Sheng, J., Yeung, T.-L., Burks, J. K., Cappello, P., Jazaeri, A. A., Kim, J.-H., Han, G. H., Birrer, M. J., Mok, S. C., & Wong, S. T. C. (2021). SIO: A Spatioimageomics Pipeline to Identify Prognostic Biomarkers Associated with the Ovarian Tumor Microenvironment. Cancers, 13(8), 1777. https://doi.org/10.3390/cancers13081777