
Role and Therapeutic Targeting of SDF-1α/CXCR4 Axis in Multiple Myeloma

Abstract
:Simple Summary
Abstract
1. Introduction
2. The Role of SDF-1α/CXCR4 in Hematopoiesis
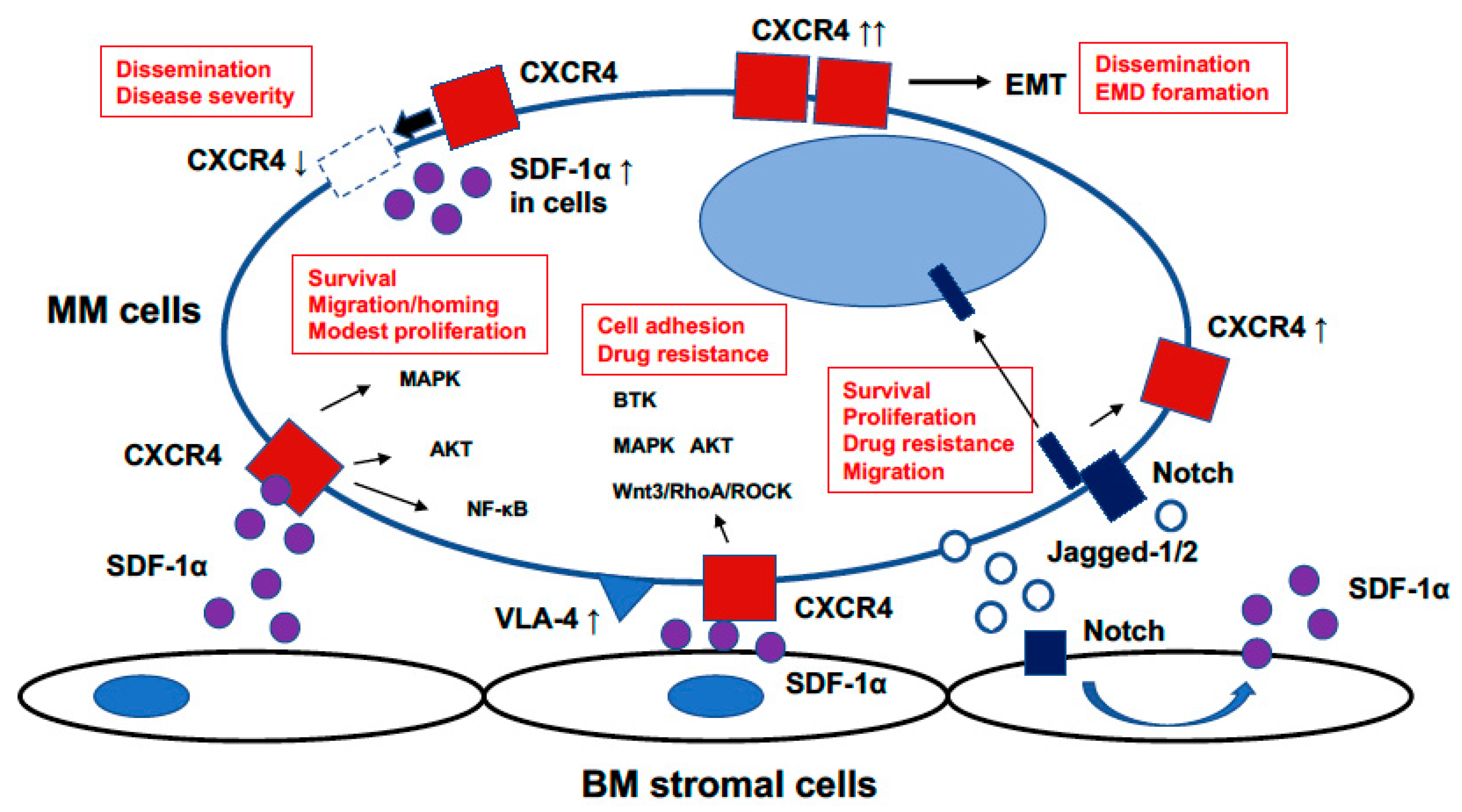
3. Role of SDF-1α/CXCR4 in MM
3.1. Roles of SDF-1α/CXCR4 on Mobilization in MM
3.2. Roles of SDF-1α/CXCR4 on Drug Resistance in MM
3.3. Notch and SDF-1α/CXCR4 in MM
3.4. SDF-1α/CXCR4 in Extramedullary Disease
4. SDF-1α/CXCR4 Targeted Therapy in MM
4.1. Plerixafor
4.2. Ulocuplumab, BMS-936564/MDX-1338
4.3. F50067
4.4. 177Lu- and 90Y-Pentixather
4.5. Olaptesed Pegol, NOX-A12

{kind=link}
| Compound | Mechanism of Action | References |
|---|---|---|
| Plerixafor | CXCR4 antagonist Inhibits migration and homing of MM cells | [101] |
| Ulocuplumab | CXCR4 antagonist Induces apoptosis in MM cells with high CXCR4 expression Inhibits SDF-1α-induced migration | [102,103] |
| F50067 | CXCR4 antagonist Inhibits cell migration and proliferation Antibody-dependent cellular cytotoxicity Compliment-dependent cytotoxicity | [104] |
| 177Lu- and 90Y-pentixather | CXCR4-directed endoradiotherapeutic agent | [108] |
| Olaptesed pegol | SDF-1 inihibitor Neutralizes SDF-1 Inhibits colonization and dissemination of MM cells | [110,111] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers. 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 1996, 184, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Toshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef]
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of CXCL12. Cell Mol. Immunol. 2018, 15, 299–311. [Google Scholar] [CrossRef]
- Kim, C.H.; Broxmeyer, H.E. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: Stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood 1998, 91, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Peled, A.; Petit, I.; Kollet, O.; Magid, M.; Ponomaryov, T.; Byk, T.; Nagler, A.; Ben-Hur, H.; Many, A.; Shultz, L.; et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 1999, 283, 845–848. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, K.; Hirota, S.; Iizasa, H.; Yoshida, H.; Kawabata, K.; Kataoka, Y.; Kitamura, Y.; Matsushima, K.; Yoshida, N.; Nishikawa, S.; et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 1998, 393, 591–594. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Kryczek, I.; Wei, S.; Keller, E.; Liu, R.; Zou, W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am. J. Physiol. Cell Physiol. 2007, 292, C987–C995. [Google Scholar] [CrossRef] [Green Version]
- Gassmann, P.; Haier, J.; Schluter, K.; Domikowsky, B.; Wendel, C.; Wiesner, U.; Kubitza, R.; Engers, R.; Schneider, S.W.; Homey, B.; et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia 2009, 11, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Ghobrial, I.M.; Roodman, G.D. Chemokines in multiple myeloma. Exp. Hematol. 2006, 34, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.K.; Pitsillides, C.M.; Spencer, J.A.; Kimlinger, T.; Ghobrial, J.M.; Jia, X.; et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood 2007, 109, 2708–2717. [Google Scholar] [CrossRef]
- Vande Broek, I.; Leleu, X.; Schots, R.; Facon, T.; Vanderkerken, K.; Van Camp, B.; Riet, I.V. Clinical significance of chemokine receptor (CCR1, CCR2 and CCR4) expression in human myeloma cells: The association with disease activity and survival. Haematologica 2006, 91, 200–206. [Google Scholar]
- Zannettino, A.C.; Farrugia, A.N.; Kortesideis, A.; Manavis, J.; To, L.B.; Martin, S.K.; Diamond, P.; Tamamura, H.; Lapidot, T.; Fujii, N.; et al. Elevated serum levels of stromal-derived factor-1alfa are associated with increased osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res. 2005, 65, 1700–1709. [Google Scholar] [CrossRef] [Green Version]
- Aiuti, A.; Webb, L.J.; Bleul, C.; Springer, T.; Gutierrez-Ramos, J.C. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J. Exp. Med. 1997, 185, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Broxmeyer, H.E.; Christopherson, K.W., II. Stromal cell-derived factor-1/CXCL12, CXCR4 and CD26 in the mobilization and homing of hematopoietic stem and progenitor cells. Curr. Med. Chem. Anti-Inflamm. Anti. Allergy Agents 2004, 3, 303–311. [Google Scholar] [CrossRef]
- Dar, A.; Kollet, O.; Lapidot, T. Mutual, reciprocal SDF-1/CXCR4 interaction between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp. Hematol. 2006, 34, 967–975. [Google Scholar] [CrossRef]
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Liles, W.C.; Broxmeyer, H.E.; Rodger, E.; Wood, B.; Hübel, K.; Cooper, S.; Hangoc, G.; Bridger, G.J.; Henson, G.W.; Calandra, G.; et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood 2004, 102, 2728–2730. [Google Scholar] [CrossRef]
- Hübel, K.; Liles, W.C.; Broxmeyer, H.E.; Rodger, E.; Wood, B.; Cooper, S.; Hangoc, G.; Macfarland, R.; Bridger, G.J.; Henson, G.W.; et al. Leukocytosis and mobilization of CD34+ hematopoietic progenitor cells by AMD3100, a CXCR4 antagonist. Support Cancer Ther. 2004, 1, 165–172. [Google Scholar] [CrossRef]
- Devine, S.M.; Flomenberg, N.; Vesole, D.H.; Liesveld, J.; Weisdorf, D.; Badel, K.; Calandra, G.; DiPersion, J.F. Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non-Hodgkin’s lymphoma. J. Clin. Oncol. 2004, 22, 1095–1102. [Google Scholar] [CrossRef]
- Grignani, G.; Perissinotto, E.; Cavalloni, G.; SChianca, F.C.; Aglietta, M. Clinical use of AMD3100 to mobilize CD34+ cells in patients affected by non-Hodgkin’s lymphoma or multiple myeloma. Clin. Oncol. 2005, 23, 3871–3872. [Google Scholar] [CrossRef]
- Liles, W.C.; Rodger, E.; Broxmeyer, H.E.; Dehner, C.; Badel, K.; Calandra, G.; Christensen, J.; Wood, B.; Price, T.H.; Dale, D.C. Augmented mobilization and collection of CD34+ hematopoietic cells from normal human volunteers stimulated with G-CSF by single-dose administration of AMD3100, a CXCR4 antagonist. Transfusion 2005, 45, 295–300. [Google Scholar] [CrossRef]
- Flomenberg, N.; Devine, S.M.; Dipersio, J.F.; Liesveld, J.L.; McCarty, J.M.; Rowley, S.D.; Vesole, D.H.; Badel, K.; Calandra, G. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 2005, 106, 1867–1874. [Google Scholar] [CrossRef]
- Papayannopoulou, T. Current mechanistic scenarios in hematopoietic stem/progenitor cell mobilization. Blood 2004, 103, 1580–1588. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, A.; Sanz-Rodriguez, F.; Rodriguez-Fernandez, J.L.; Albella, B.; Blaya, C.; Wright, N.; Cabanas, C.; Prosper, F.; Gutierrez-Ramos, J.C.; Teixido, J. Chemokine stromal cell-derived factor-1α modulates VLA-4 integrin-dependent adhesion to fibronectin and VCAM-1 on bone marrow hematopoietic progenitor cells. Exp. Hematol. 2001, 29, 345–355. [Google Scholar] [CrossRef]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Podar, K.; Akiyama, M.; Gupta, D.; Richardson, P.; Munshi, N.; Anderson, K.C. The biological sequelae of stromal cell-derived factor-1α in multiple myeloma. Mol. Cancer Ther. 2002, 1, 539–544. [Google Scholar]
- Hideshima, T.; Chauhan, D.; Schlossman, R.; Richardson, P.; Anderson, K.C. The role of tumor necrosis factor α in the pathogenesis of human multiple myeloma: Therapeutic applications. Oncogene 2001, 20, 4519–4527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliot, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Kijowski, J.; Baj-Krzyworzeka, M.; Majka, M.; Reca, R.; Marquez, L.A.; Christofidou-Solomidou, M.; Janowaska-Wieczorek, A.; Ratajczak, M.Z. The SDF-1-CXCR4 axis stimulates VEGF secretion and activates integrins but does not affect proliferation and survival in lymphohematopoietic cells. Stem Cells 2001, 19, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Wang, J.; Zhang, X.; Zhang, J.J.; Wu, F.; Pang, Y.; Zhong, Y.; Wang, J.; Wang, W.; Lyu, X.; et al. Single-cell RNA sequencing reveals chemokine self-feeding of myeloma cells promotes extramedullary metastasis. FEBS Lett. 2020, 594, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.K.; Dewar, A.L.; Farrugia, A.N.; Horvath, N.; Gronthos, S.; To, L.B.; Zannettino, A.C. Tumor angiogenesis is associated with plasma levels of stromal-derived factor-1-α in patients with multiple myeloma. Clin. Cancer Res. 2006, 12, 6973–6977. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, S.; Prada, C.P.; Richardson, P.G. How I treat the young patient with multiple myeloma. Blood 2018, 132, 1114–1124. [Google Scholar] [CrossRef]
- Vora, A.J.; Toh, C.H.; Peel, J.; Greaves, M. Use of granulocyte colony-stimulating factor (G-CSF) for mobilizing peripheral blood stem cells: Risk of mobilizing clonal myeloma cells in patients with bone marrow infiltration. Br. J. Haematol. 1994, 86, 180–182. [Google Scholar] [CrossRef]
- Keklik, M.; Karakus, E.; Kaynar, L.; Akyol, G.; Guven, Z.T.; Celik, S.; Baydar, M.; Sanlı, N.; Unal, A.; Cetin, M. Low-dose cyclophosphamide and granulocyte colony-stimulating factor are sufficient for peripheral blood stem cell mobilization in patients with multiple myeloma. Transfus. Apher. Sci. 2020, 59, 102844. [Google Scholar] [CrossRef]
- Lane, T.A.; Law, P.; Maruyama, M.; Young, D.; Burgess, J.; Mullen, M.; Mealiffe, M.; Terstappen, L.W.; Hardwick, A.; Moubayed, M.; et al. Harvesting and enrichment of HPC mobilized into the peripheral blood of normal donors by GM-CSF or G-CSF: Potential role in allogenic transplantation. Blood 1995, 85, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Peters, W.P.; Rosner, G.; Ross, M.; Vredenburgh, J.; Meisenberg, B.; Gilbert, C.; Kurtzberg, J. omparative effect of GM-CSF and G-CSF on priming of peripheral blood progenitor cells for use with autologous bone marrow after high dose chemotherapy. Blood 1993, 81, 1709–1719. [Google Scholar]
- DiPersio, J.F.; Stadtmauer, E.A.; Nademanee, A.; Micallef, I.N.; Stiff, P.J.; Kaufman, J.L.; Maziarz, R.T.; Hosing, C.; Früehauf, S.; Horwitz, M.; et al. Plerixor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cell for autologous stem cell transplantation in patients with multiple myeloma. Blood 2009, 113, 5720–5726. [Google Scholar] [CrossRef]
- Dreyfus, F.; Ribrag, V.; Leblond, V.; Ravaud, P.; Melle, J.; Quarre, M.C.; Pillier, C.; Boccaccio, C.; Varet, B. Detection of malignant B cells in peripheral blood stem cell collections after chemotherapy in patients with multiple myeloma. Bone Marrow Transpl. 1995, 15, 707–712. [Google Scholar]
- Gazitt, Y.; Tian, E.; Barlogie, B.; Reading, C.L.; Vesole, D.H.; Jagannath, S.; Schnell, J.; Hoffman, R.; Tricot, G. Differential mobilization of myeloma cells and normal hematopoietic stem cells in multiple myeloma after treatment with cyclophosphamide and GM-CSF. Blood 1996, 87, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Gazitt, Y.; Reading, C. Autologous transplantation with tumor-free graft: A model for multiple myeloma patients. Leuk Lymphoma 1996, 27, 202–212. [Google Scholar] [CrossRef]
- Tricot, G.; Gazitt, Y.; Leemhuis, T.; Jagannath, S.; Desikan, K.R.; Siegel, D.; Fassas, A.; Tindle, S.; Nelson, J.; Juttner, C.; et al. Collection, engraftment kinetics and tumor contamination of highly purified hematopoietic progenitor cells to support high dose therapy in multiple myelomas. Blood 1998, 91, 4489–4495. [Google Scholar] [CrossRef]
- Gazitt, Y.; Akay, C. Mobilization of myeloma cells involves SDF-1/CXCR4 signaling and downregulation of VLA-4. Stem Cells 2004, 22, 65–73. [Google Scholar] [CrossRef]
- Nahi, H.; Celanovic, M.; Liu, Q.; Lund, J.; Peceliunas, V. A Pilot, Exploratory, Randomized, Phase II Safety Study Evaluating Tumor Cell Mobilization and Apheresis Product Contamination in Patients Treated with Granulocyte Colony-Stimulating Factor Alone or Plus Plerixafor. Biol. Blood Marrow Transpl. 2019, 25, 34–40. [Google Scholar] [CrossRef]
- Kopp, H.G.; Yildirim, S.; Weisel, K.C.; Kanz, L.; Vogel, W. Contamination of autologous peripheral blood progenitor cell grafts predicts overall survival after high-dose chemotherapy in multiple myeloma. J. Cancer Res. Clin. Oncol. 2009, 135, 637–642. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef]
- Waldschmidt, J.M.; Simon, A.; Wider, D.; Muller, S.J.; Follo, M.; Ihorst, G.; Decker, S.; Lorenz, J.; Chatterjee, M.; Azab, A.K.; et al. CXCL12 and CXCR7 are relevant targets to reverse cell adhesion-mediated drug resistance in multiple myeloma. Br. J. Haematol. 2017, 179, 36–49. [Google Scholar] [CrossRef]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Dehghanifard, A.; Kaviani, S.; Abroun, S.; Mehdizadeh, M.; Saiedi, S.; Maali, A.; Ghaffari, S.; Azad, M. Various signaling pathways in multiple myeloma cells and effects of treatment on these pathways. Clin. Lymphoma Myeloma Leuk 2018, 18, 311–320. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Damiano, J.S.; Buyuksal, I.; Pledger, W.J.; Dalton, W.S. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 2000, 19, 4319–4327. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, H.M.; Lv, Y.Q.; Tang, S.M.; Cheng, P. Blockade of SDF-1/CXCR4 reduces adhesion-mediated chemoresistance of multiple myeloma cells via interacting with interleukin-6. J. Cell Physiol. 2019, 234, 19702–19714. [Google Scholar] [CrossRef]
- Sun, L.; Liu, L.L.; Liu, X.Q.; Wang, Y.F.; Li, M.B.; Yao, L.P.; Yang, J.J.; Ji, G.L.; Guo, C.C.; Pan, Y.L.; et al. MGr1- Ag/37LRP induces cell adhesion-mediated drug resistance through FAK/PI3K and MAPK pathway in gastric cancer. Cancer Sci. 2014, 105, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Kobune, M.; Chiba, H.; Kato, J.; Kato, K.; Nakamura, K.; Kawano, Y.; Takada, K.; Takimoto, R.; Takayama, T.; Hamada, H.; et al. Wnt3/RhoA/ROCK signaling pathway is involved in adhesion-mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol. Cancer Ther. 2007, 6, 1774–1784. [Google Scholar] [CrossRef] [Green Version]
- Schmidmaier, R.; Baumann, P.; Simsek, M.; Day-yani, F.; Emmerich, B.; Meinhardt, G. The HMG-CoA reductase inhibitor simvastatin overcomes cell adhesion-mediated drug resistance in multiple myeloma by geranylgeranylation of Rho protein and activation of Rho kinase. Blood 2004, 104, 1825–1832. [Google Scholar] [CrossRef]
- Bam, R.; Venkateshaiah, S.U.; Khan, S.; Ling, W.; Randal, S.S.; Li, X.; Zhang, Q.; van Rhee, F.; Barlogie, B.; Epstein, J.; et al. Role of Bruton’s tyrosine kinase (BTK) in growth and metastasis of INA6 myeloma cells. Blood Cancer J. 2014, 4, e234. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shi, J.; Gu, Z.; Salama, M.E.; Das, S.; Wen-dlandt, E.; Xu, H.; Huang, J.; Tao, Y.; Hao, M.; et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015, 75, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.S.; Chang, B.Y.; Chang, S.; Tong, T.; Ham, S.; Sherry, B.; Burger, J.A.; Rai, K.R.; Chiorazzi, N. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia 2016, 30, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bam, R.; Ling, W.; Khan, S.; Pennisi, A.; Venkateshaiah, S.U.; Li, X.; van Rhee, F.; Usmani, S.; Barlogie, B.; Shaughnessy, J.; et al. Role of Bruton’s tyrosine kinase in myeloma cell migration and induction of bone disease. Am. J. Hematol. 2013, 88, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wei, R.; Liu, S.; Qiao, L.; Hou, J.; Gu, C.; Yang, Y. BTK induces CAM-DR through regulation of CXCR4 degradation in multiple myeloma. Am. J. Transl. Res. 2019, 11, 4139–4150. [Google Scholar] [PubMed]
- Richardson, P.G.; Hungria, V.T.M.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Guenther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: Outcomes by prior treatment. Blood 2016, 127, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Chari, A.; Cho, H.J.; Dhadwal, A.; Morgan, G.; La, L.; Zarychta, K.; Catamero, D.; Florendo, E.; Stevens, N.; Verina, D.; et al. A phase 2 study of panobinostat with lenalidomide and weekly dexamethasone in myeloma. Blood Adv. 2017, 1, 1575–1583. [Google Scholar] [CrossRef] [Green Version]
- Beider, K.; Bitner, H.; Voevoda-Dimenshtein, V.; Rosenberg, E.; Sirovsky, Y.; Magen, H.; Canaani, J.; Ostrovsky, O.; Shilo, N.; Shimoni, A.; et al. The mTOR inhibitor everolimus overcomes CXCR4-mediated resistance to histone deacetylase inhibitor panobinostat through inhibition of p21 and mitotic regulators. Biochem. Pharmacol. 2019, 168, 412–428. [Google Scholar] [CrossRef]
- Osborne, B.; Miele, L. Notch and the immune system. Immunity 1999, 11, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, L.; Comi, P.; Cobos, E.; Kast, W.M.; Chiriva-Internati, M.; Chiaramonte, R. Notch-ing from T-cell to B-cell ly,mphoid malignancies. Cancer Lett. 2011, 308, 1–13. [Google Scholar] [CrossRef]
- Skrtic, A.; Korac, P.; Krito, D.R.; Ajdukovic Stojisavljevic, R.; Ivankovic, D.; Dominis, M. Immunohistochemical analysis of NOTCH1 and JAGGED1 expression in multiple myeloma and monoclonal gammopathy of undetermined significance. Hum. Pathol. 2010, 41, 1702–1710. [Google Scholar] [CrossRef]
- Ghoshal, P.; Nganga, A.J.; Moran-Giuati, J.; Szafranek, A.; Johnson, T.R.; Bigelow, A.J.; Houde, C.M.; Avet-Loiseu, H.; Smiraglia, D.J.; Chanan-khan, N.E.A.A.; et al. Loss of the SMRT/NCorR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009, 69, 4380–4383. [Google Scholar] [CrossRef] [Green Version]
- Jundt, F.; Probsting, K.S.; Anagnostopoulos, I.; Muehlinghaus, G.; Chaterjee, M.; Mathas, S.; Bargou, R.C.; Manz, R.; Stein, H.; Dorken, B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 2004, 103, 3511–3515. [Google Scholar] [CrossRef]
- Hedvat, C.V.; Comenzo, R.L.; Teruya-Feldsein, J.; Olshen, A.B.; Ely, S.A.; Osman, K.; Zhang, Y.; Kalalonda, N.; Nimer, S.D. Insights into extramedullary tumor cell growth revealed by expression profiling of human plasmacytomas and multiple myeloma. Br. J. Haematol. 2003, 122, 728–744. [Google Scholar] [CrossRef]
- Houde, C.; Li, Y.; Song, L.; Barton, K.; Zhang, Q.; Godwin, J.; Nand, S.; Toor, A.; Alkan, S.; Smadja, N.V.; et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood 2004, 104, 3697–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Adachi, Y.; Ohtsuki, Y. Skeletophin, a novel ubiquitin ligase to the intracellular region of Jagged-2, is aberrantly expressed in multiple myeloma. Am. J. Pathol. 2005, 166, 1817–1826. [Google Scholar] [CrossRef] [Green Version]
- Nefedova, Y.; Cheng, P.; Alsina, M.; Dalton, D.I. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood 2004, 103, 3503–3510. [Google Scholar] [CrossRef]
- Mirandola, L.; Chiriva-Internati, M.; Montagna, D.; Locatelli, F.; Zecca, M.; Ranzani, M.; Basile, A.; Locati, M.; Cobos, E.; Kast, W.M.; et al. Notch1 regulates chemotaxis and proliferation by controlling the chemokine receptors 5 and 9 in T-cell acute lymphoblastic leukemia. J. Pathol. 2011, 7, 713–722. [Google Scholar]
- Schwarzer, R.; Kaiser, M.; Acikgoz, O.; Heider, U.; Mathas, S.; Preissner, R.; Sezer, O.; Doerken, B.; Jundt, F. Notch inhibition blocks multiple myeloma cell-induced osteoclast activation. Leukemia 2008, 22, 2273–2277. [Google Scholar] [CrossRef]
- Mirandola, L.; Apicella, L.; Colombo, M.; Yu, Y.; Latonova, N.; Lazzari, E.; Lancellotti, M.; Bulfamante, G.; Cobos, E.; Chiriva-Internati, M.; et al. Anti-Notch treatment prevents multiple myeloma cells localization of the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia 2013, 27, 1558–1566. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Garavelli, S.; Mazzola, M.; Platonova, N.; Giannandrea, D.; Colella, R.; Apicells, L.; Lancellotti, M.; Lesma, E.; Ancona, S.; et al. Multiple myeloma exploits Jagged1 and Jagged2 to promote intrinsic and bone marrow-dependent drug resistance. Haematologica 2020, 105, 1925–1936. [Google Scholar] [CrossRef] [Green Version]
- Blade, J.; Lust, J.; Kyle, R.A. Immunoglobulin D multiple myeloma: Presenting features, response to therapy, and survival in a series of 53 cases. J. Clin. Oncol. 1994, 12, 2398–2402. [Google Scholar] [CrossRef]
- Blade, J.; Kyle, R.A.; Greipp, P.R. Presenting features and prognosis in 72 patients with multiple myeloma who were younger than 40 years. Br. J. Haematol. 1996, 93, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Davies, F.E.; Boyd, K.; Thomas, K.; Dines, S.; Saso, R.M.; Potter, M.N.; Ethell, M.E.; Shaw, B.E.; Morgan, G.J. The impact of extramedullary disease at presentation in the outcome of myeloma. Leuk Lymphoma 2009, 50, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Varettoni, M.; Corso, A.; Pica, G.; Mangiacavalli, S.; Pascutto, C.; Lazzarino, M. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma. A longitudinal study on 1003 consecutive patients. Ann. Oncol. 2009, 21, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Blade, J.; Fernandez de Larrea, C.; Rosinol, L.; Cibeira, M.T.; Jimenez, R.; Powles, R. Soft-tissue plasmacytomas in multiple myeloma: Incidence, mechanisms of extramedullary spread, and treatment approach. J. Clin. Oncol. 2011, 29, 380–3821. [Google Scholar] [CrossRef]
- Lee, S.E.; Kim, J.H.; Jeon, Y.W.; Yoon, J.H.; Shin, S.H.; Eom, K.S.; Kim, Y.J.; Kim, H.J.; Lee, S.; Cho, S.G.; et al. Impact of extramedullary plasmacytomas on outcomes according to treatment approach in newly diagnosed symptomatic multiple myeloma. Ann. Hematol. 2015, 94, 445–452. [Google Scholar] [CrossRef]
- Molica, S. Extramedullary involvement: An emerging problem in multiple myeloma. Clin. Adv. Hematol. Oncol. 2012, 10, 268–269. [Google Scholar]
- Kakati, B.R.; Krishna, K.; Krishna, S.G.; Sharma, S.G.; Sanathkumar, N.; Rego, R.F. Extensive extramedullary disease involving the colon in multiple myeloma: A case report and review of literature. J. Gastrointest Cancer 2012, 43, 379–381. [Google Scholar] [CrossRef]
- Brade, J.; de Larrea, C.F.; Rosinol, L. Extramedullary involvement in multiple myeloma. Haematologica 2012, 97, 1618–1619. [Google Scholar]
- Oliveira, A.M.; Maria, D.A.; Metzger, M.; Linardi, C.; Giorgi, R.R.; Moura, F.; Martinez, G.A.; Bydlowski, S.P.; Novak, E.M. Thalidomide treatment down-regulates SDF-1α and CXCR4 expression in multiple myeloma patients. Leuk. Res. 2009, 33, 970–973. [Google Scholar] [CrossRef]
- Stessman, H.; Mansoor, A.; Zhan, F.; Janz, S.; Linden, M.A.; Baughn, L.B.; Van Ness, B. Reduced CXCR4 expression is associated with extramedullary disease in a mouse model of myeloma and predicts poor survival in multiple myeloma patients treated with bortezomib. Leukemia 2013, 27, 2075–2077. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar] [CrossRef]
- Shaughnessy, J.D., Jr.; Qu, P.; Usmani, S.; Heuck, C.J.; Zhang, Q.; Zhou, Y.; Tian, E.; Hanamura, I.; van Rhee, F.; Anaissie, E.; et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with total therapy 3. Blood 2011, 118, 3512–3524. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Danoff, T.M.; Kalluri, R.; Neilson, E.G. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997, 273, F563–F574. [Google Scholar] [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 11, 335–348. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep. 2015, 12, 622–635. [Google Scholar]
- Ghobrial, I.M.; Liu, C.J.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosesitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 2019, 94, 1244–1253. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Liu, C.J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.; et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Fouquet, G.; Guidez, S.; Richez, V.; Stoppa, A.M.; Le Tourneau, C.; Macro, M.; Gruchet, C.; Bobin, A.; Moya, N.; Syshenko, T.; et al. Phase I dose-escalation study of F50067, a humanized anti-CXCR4 monoclonal antibody alone and in combination with lenalidomide and low-dose dexamethasone, in relapsed or refractory multiple myeloma. Oncotarget 2018, 9, 23890–23899. [Google Scholar] [CrossRef] [PubMed]
- Demmer, O.; Gourni, E.; Schumacher, U.; Kessler, H.; Wester, H.J. PET imaging of CXCR4 receptors in cancer by a new optimized ligand. ChemMedChem 2011, 6, 1789–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourni, E.; Demmer, O.; Schottelius, M.; D’Alessandria, C.; Schulz, S.; Dijkgraaf, I.; Schumacher, U.; Schwaiger, M.; Kessler, H.; Wester, H.J. PET of CXCR4 expression by a 68Ga-labeled highly specific targeted contrast agent. J. Nucl. Med. 2011, 52, 1803–1810. [Google Scholar] [CrossRef] [Green Version]
- Philipp-Abbrederis, K.; Herrmann, K.; Knop, S.; Schottelius, M.; Eiber, M.; Lückerath, K.; Pietschmann, E.; Habringer, S.; Gerngroß, C.; Franke, K.; et al. In vivo molecular imaging of chemokine receptor CXCR4 expression in patients with advanced multiple myeloma. EMBO Mol. Med. 2015, 7, 477–487. [Google Scholar] [CrossRef]
- Herrmann, K.; Schottelius, M.; Lapa, C.; Osl, T.; Poschenrieder, A.; Hänscheid, H.; Lückerath, K.; Schreder, M.; Bluemel, C.; Knott, M.; et al. First-in-human experience of CXCR4-directed endoradiotherapy with 177Lu- and 90Y-labeled pentixather in advanced-stage multiple myeloma with extensive intra- and extramedullary disease. J. Nucl. Med. 2016, 57, 248–251. [Google Scholar] [CrossRef] [Green Version]
- Bouyssou, J.M.; Ghobrial, I.M.; Roccaro, A.M. Targeting SDF-1 in multiple myeloma tumor microenvironment. Cancer Lett. 2015, 380, 315–318. [Google Scholar] [CrossRef]
- Roccaro, A.; Sacco, A.; Purschke, W.G.; Moschetta, M.; Buchner, K.; Maasch, C.; Aboralski, D.; Zollner, S.; Vonhoff, S.; Mishima, Y.; et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014, 9, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, H.; Weisel, K.; Petrucci, M.T.; Leleu, X.; Cafro, A.M.; Garderet, L.; Leitgeb, C.; Foa, R.; Greil, R.; Yakoub-Agha, I.; et al. Olaptesed pegol, an anti-CXCL12/SDF-1 Spiegelmer, alone and with bortezomib-dexamethasone in relapsed/refractory multiple myeloma: A phase IIa study. Leukemia 2017, 31, 997–1000. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, S.; Sato, T.; Maeta, T. Role and Therapeutic Targeting of SDF-1α/CXCR4 Axis in Multiple Myeloma. Cancers 2021, 13, 1793. https://doi.org/10.3390/cancers13081793
Ito S, Sato T, Maeta T. Role and Therapeutic Targeting of SDF-1α/CXCR4 Axis in Multiple Myeloma. Cancers. 2021; 13(8):1793. https://doi.org/10.3390/cancers13081793
Chicago/Turabian StyleIto, Shigeki, Tsuyoshi Sato, and Takahiro Maeta. 2021. "Role and Therapeutic Targeting of SDF-1α/CXCR4 Axis in Multiple Myeloma" Cancers 13, no. 8: 1793. https://doi.org/10.3390/cancers13081793