
A Targeted-Covalent Inhibitor of 17β-HSD1 Blocks Two Estrogen-Biosynthesis Pathways: In Vitro (Metabolism) and In Vivo (Xenograft) Studies in T-47D Breast Cancer Models



Abstract
:Simple Summary
Abstract
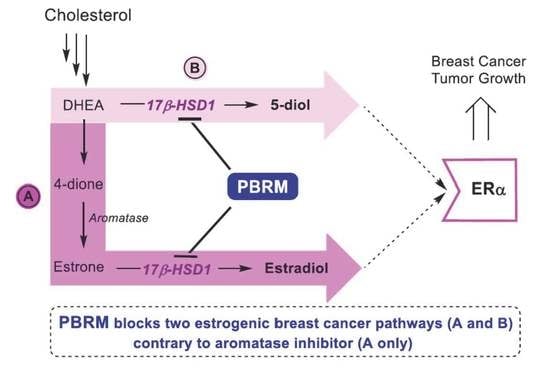
1. Introduction
2. Results
2.1. DHEA as Precursor of Estrogenic Effects
2.2. E1 as Precursor of Estrogenic Effects
2.3. Assessment of PBRM Frequency of Administration
3. Discussion
4. Materials and Methods
4.1. Inhibitors
4.2. In Vitro Studies
4.2.1. Cell Culture
4.2.2. Metabolism of DHEA in T-47D Cells
4.2.3. 17β-HSD1 Inhibition Assays
4.3. In Vivo Studies (T-47D Xenografts in Nude Mice)
4.3.1. Animals for Xenografts
4.3.2. Stimulation of T-47D Tumor Growth by DHEA in Nude OVX Mice
4.3.3. Inhibition of DHEA-Stimulated T-47D Tumor Growth in Nude OVX Mice
4.3.4. Inhibition of E1-Stimulated T-47D Tumor Growth in Nude OVX Mice (Increased Dose of PBRM)
4.3.5. Inhibition of E1-Stimulated T-47D Tumor Growth in Nude OVX Mice (Frequency of PBRM Administration)
4.4. Dosage of Steroids and PBRM
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Bush, N.J. Advances in hormonal therapy for breast cancer. Semin. Oncol. Nurs. 2007, 23, 46–54. [Google Scholar] [CrossRef]
- MacGregor, J.I.; Jordan, V.C. Basic guide to the mechanism of antiestrogen action. Pharmacol. Rev. 1998, 50, 151–196. [Google Scholar]
- Maximov, P.Y.; Lee, T.M.; Jordan, V.C. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr. Clin. Pharmacol. 2013, 8, 135–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Adamo, V.; Iorfida, M.; Montalto, E.; Festa, V.; Garipoli, C.; Scimone, A.; Zanghi, M.; Caristi, N. Overview and new strategies in metastatic breast cancer (MBC) for treatment of tamoxifen-resistant patients. Ann. Oncol. 2007, 18 (Suppl. 6), 53–57. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2006, 25, 947–970. [Google Scholar] [CrossRef]
- Poirier, D. New cancer drugs targeting the biosynthesis of estrogens and androgens. Drug Dev. Res. 2008, 69, 304–318. [Google Scholar] [CrossRef]
- Bruno, R.D.; Njar, V.C.O. Targeting cytochrome P450 enzymes: A new approach in anti-cancer drug development. Bioorg. Med. Chem. 2007, 15, 5047–5060. [Google Scholar] [CrossRef]
- Foster, P.A. Steroid metabolism in breast cancer. Minerva Endocrinol. 2008, 33, 27–37. [Google Scholar]
- Subramanian, A.; Salhab, M.; Mokbel, K. Oestrogen producing enzymes and mammary carcinogenesis: A review. Breast Cancer Res. Treat. 2008, 111, 191–202. [Google Scholar] [CrossRef]
- Smith, H.J.; Nicholls, P.J.; Simons, C.; Le Lain, R. Inhibitors of steroidogenesis as agents for the treatment of hormone-dependent cancers. Exp. Opin. Ther. Pat. 2001, 11, 789–824. [Google Scholar] [CrossRef]
- Brueggemeier, R.W.; Hackett, J.C.; Diaz-Cruz, E.S. Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 2005, 26, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J. Aromatase inhibitors for breast cancer: Exemestane (Aromasin), anastrozole (Arimidex) and letrozole (Femara). In The Art of Drug Synthesis; Johnson, D.S., Li, J.J., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 31–38. [Google Scholar]
- Potter, B.V.L. Steroid sulphatase inhibition via aryl sulphamates: Clinical progress, mechanism and future prospects. J. Mol. Endocrinol. 2018, 61, T233–T252. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Singh, J.; Singh, D.; Singh Jaggi, A.; Singh, N. Sulfatase inhibitors for recidivist breast cancer treatment: A chemical review. Eur. J. Med. Chem. 2016, 114, 170–190. [Google Scholar] [CrossRef]
- Costa, E.V.; Sousa, E.; Choosang, K.; Singh, S.; Rocha, J.; Lima, R.T.; Pakkong, P.; Ahmed, S.; Vasconcelos, M.H.; Montanari, C.A.; et al. Structure based design, synthesis, and evaluation of potential inhibitors of steroid sulfatase. Curr. Top. Med. Chem. 2014, 14, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, Y.A.; Taylor, S.D. Steroid derivatives as inhibitors of steroid sulfatase. J. Steroid Biochem. Mol. Biol. 2013, 137, 183–198. [Google Scholar] [CrossRef]
- Purohit, A.; Foster, P.A. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J. Endocrinol. 2012, 212, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltais, R.; Poirier, D. Steroid sulfatase inhibitors: A review covering the promising 2000–2010 decade. Steroids 2011, 76, 929–948. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Potter, B.V.L. Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women’s health. J. Med. Chem. 2015, 58, 7634–7658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombes, C.; Cardoso, F.; Isambert, N.; Lesimple, T.; Soulié, P.; Peraire, C.; Fohanno, V.; Kornowski, A.; Ali, T.; Schmid, P. A phase I dose escalation study to determine the optimal biological dose of irosustat, an oral steroid sulfatase inhibitor, in postmenopausal women with estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2013, 140, 73–82. [Google Scholar] [CrossRef]
- Palmieri, C.; Stein, R.C.; Liu, X.; Hudson, E.; Nicholas, H.; Sasano, H.; Guestini, F.; Holcombe, C.; Barrett, S.; Kenny, L.; et al. IRIS trial participants. IRIS study: A phase II study of the steroid sulfatase inhibitor Irosustat when added to an aromatase inhibitor in ER-positive breast cancer patients. Breast Cancer Res. Treat. 2017, 165, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, A. 17Beta-hydroxysteroid dehydrogenase enzymes and breast cancer. J. Steroid Biochem. Mol. Biol. 2009, 114, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Ando, A.; Shiba, E.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Involvement of up-regulation of 17beta-hydroxysteroid dehydrogenase type 1 in maintenance of intratumoral high estradiol levels in postmenopausal breast cancers. Int. J. Cancer. 2001, 94, 685–689. [Google Scholar] [CrossRef]
- Labrie, F.; Luu-The, V.; Lin, S.X.; Labrie, C.; Simard, J.; Breton, R.; Bélanger, A. The key role of 17β-hydroxysteroid dehydrogenases in sex steroid biology. Steroids 1997, 62, 148–158. [Google Scholar] [CrossRef]
- Suzuki, T.; Miki, Y.; Nakamura, Y.; Moriya, T.; Ito, K.; Ohuchi, N.; Sasano, H. Sex steroid-producing enzymes in human breast cancer. Endocr. Relat. Cancer 2005, 12, 701–720. [Google Scholar] [CrossRef] [Green Version]
- Lanisnik Rizner, T. Estrogen biosynthesis, phase I and phase II metabolism, and action in endometrial cancer. Mol. Cell. Endocrinol. 2013, 381, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, W.Q.; Lin, S.X. Interaction of androst-5-ene-3β,17β-diol and 5α-androstane-3β,17β-diol with estrogen and androgen receptors: A combined binding and cell study. J. Steroid Biochem. Mol. Biol. 2013, 137, 316–321. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Tutill, H.J.; Purohit, A. 17β-hydroxysteroid dehydrogenase inhibitors. Minerva Endocrinol. 2010, 35, 87–108. [Google Scholar]
- Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Poirier, D. 17beta-Hydroxysteroid dehydrogenase inhibitors: A patent review. Expert Opin. Ther. Pat. 2010, 20, 1123–1145. [Google Scholar] [CrossRef] [PubMed]
- Poirier, D. Recent advances in development of inhibitors of 17β-hydroxysteroid dehydrogenases. Anti-Cancer Agents Med. Chem. 2009, 9, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Tutill, H.J.; Purohit, A.; Reed, M.J. Design and validation of specific inhibitors of 17beta-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr. Relat. Cancer 2008, 15, 665–692. [Google Scholar] [CrossRef] [Green Version]
- Brozic, P.; Rizner, T.L.; Gobec, S. Inhibitors of 17β-hydroxysteroid dehydrogenase type 1. Curr. Med. Chem. 2008, 15, 137–150. [Google Scholar]
- Fournier, D.; Poirier, D.; Mazumdar, M.; Lin, S.X. Design and synthesis of bisubstrate inhibitors of type 1 17β-hydroxysteroid dehydrogenase: Overview and perspectives. Eur. J. Med. Chem. 2008, 43, 2298–2306. [Google Scholar] [CrossRef]
- Poirier, D. Inhibitors of 17 beta-hydroxysteroid dehydrogenases. Curr. Med. Chem. 2003, 10, 453–477. [Google Scholar] [CrossRef]
- Lin, S.X.; Poirier, D.; Adamski, J. A challenge for medicinal chemistry by the 17β-hydroxysteroid dehydrogenase superfamily: An integrated biological function and inhibition study. Curr. Top. Med. Chem. 2013, 13, 1164–1174. [Google Scholar] [CrossRef]
- National Institutes of Health U.S. National Library of Medicine. A Study to Investigate the Safety, Tolerability, Food Effect, Pharmacokinetics and Pharmacodynamics of FOR-6219. Available online: https://clinicaltrials.gov/ct2/show/NCT03709420 (accessed on 7 January 2021).
- Maltais, R.; Ayan, D.; Poirier, D. Crucial role of 3-bromoethyl in removing the estrogenic activity of 17beta-HSD1 inhibitor 16beta-(m-carbamoylbenzyl) estradiol. ACS Med. Chem. Lett. 2011, 2, 678–681. [Google Scholar] [CrossRef] [Green Version]
- Ayan, D.; Maltais, R.; Roy, J.; Poirier, D. A new nonestrogenic steroidal inhibitor of 17beta-hydroxysteroid dehydrogenase type I blocks the estrogen-dependent breast cancer tumor growth induced by estrone. Mol. Cancer Ther. 2012, 11, 2096–2104. [Google Scholar] [CrossRef] [Green Version]
- Maltais, R.; Ayan, D.; Trottier, A.; Barbeau, X.; Lagüe, P.; Bouchard, J.E.; Poirier, D. Discovery of a non-estrogenic irreversible inhibitor of 17beta-hydroxysteroid dehydrogenase type 1 from 3-substituted-16-beta-(m-carbamoylbenzyl)-estradiol derivatives. J. Med. Chem. 2014, 57, 204–222. [Google Scholar] [CrossRef]
- Trottier, A.; Maltais, R.; Ayan, D.; Barbeau, X.; Roy, J.; Perreault, M.; Poulin, R.; Lagüe, P.; Poirier, D. Insight into the mode of action and selectivity of PBRM, a covalent steroidal inhibitor of 17β-hydroxysteroid dehydrogenase type 1. Biochem. Pharmacol. 2017, 144, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Maltais, R.; Poirier, D.; Lin, S.X. Combined biophysical chemistry reveals a new covalent inhibitor with a low-reactivity alkyl halide. J. Phys. Chem. Lett. 2018, 9, 5275–5280. [Google Scholar] [CrossRef] [PubMed]
- Luu-The, V. Assessment of steroidogenesis and steroidogenic enzyme functions. J. Steroid Biochem. Mol. Biol. 2013, 137, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Forney, J.P.; Milewich, L.; Chen, G.T.; Garlock, J.L.; Schwarz, B.E.; Edman, C.D.; MacDonald, P.C. Aromatization of androstenedione to estrone by human adipose tissue in vitro. Correlation with adipose tissue mass, age, and endometrial neoplasia. J. Clin. Endocrinol. Metab. 1981, 53, 192–199. [Google Scholar] [CrossRef]
- Poulin, R.; Labrie, F. Stimulation of cell proliferation and estrogenic response by adrenal C19-delta5-steroids in the ZR-75-1 human breast cancer cell line. Cancer Res. 1986, 46, 4933–4937. [Google Scholar] [PubMed]
- Aspinall, S.R.; Stamp, S.; Davison, A.; Shenton, B.K.; Lennard, T.W.J. The proliferative effect of 5-androstene-3 beta,17 beta-diol and 5 alpha-dihydrotestosterone on cell cycle analysis and cell proliferation in MCF7, T47D and MDAMB231 breast cancer cell lines. J. Steroid Biochem. Mol. Biol. 2004, 88, 37–51. [Google Scholar] [CrossRef]
- Laplante, Y.; Rancourt, C.; Poirier, D. Relative involvement of three 17β-hydroxysteroid dehydrogenases (types 1, 7 and 12) in the formation of estradiol in various breast cancer cell lines using selective inhibitors. Mol. Cell. Endocrinol. 2009, 301, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Ryde, C.M.; McNicholls, J.E.; Dowsett, M. Steroid and growth factor modulation of aromatase activity in MCF7 and T47D breast carcinoma cell lines. Cancer Res. 1992, 52, 1411–1415. [Google Scholar]
- Sadekova, S.I.; Tan, L.; Chow, T.Y.K. Identification of the aromatase in the breast carcinoma cell lines T47D and MCF-7. Anticancer Res. 1994, 14, 507–512. [Google Scholar]
- Laplante, Y.; Cadot, C.; Fournier, M.A.; Poirier, D. Estradiol and estrone C-16 derivatives as inhibitors of type 1 17beta-hydroxysteroid dehydrogenase: Blocking of ER+ breast cancer cell proliferation induced by estrone. Bioorg. Med. Chem. 2008, 16, 1849–1860. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Jost, C.; Nitsche, C.; Scholz, T.; Roux, L.; Klein, C.D. Promiscuity and selectivity in covalent enzyme inhibition: A systematic study of electrophilic fragments. J. Med. Chem. 2014, 57, 7590–7599. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Messinger, J.; Husen, B.; Koskimies, P.; Hirvelä, L.; Kallio, L.; Saarenketo, P.; Thole, H. Estrone C15 derivatives—A new class of 17beta-hydroxysteroid dehydrogenase type 1 inhibitors. Mol. Cell. Endocrinol. 2009, 301, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.M.; Foster, P.A.; Tutill, H.J.; Parsons, M.F.C.; Newman, S.P.; Chander, S.K.; Allan, G.M.; Lawrence, H.R.; Vicker, N.; Potter, B.V.L.; et al. 17β-Hydroxysteroid dehydrogenase type 1, and not type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int. J. Cancer 2008, 122, 1931–1940. [Google Scholar] [CrossRef]
- Mitropoulou, T.N.; Tzanakakis, G.N.; Kletsas, D.; Kalofonos, H.P.; Karamanos, N.K. Letrozole as a potent inhibitor of cell proliferation and expression of metalloproteinases (MMP-2 and MMP-9) by human epithelial breast cancer cells. Int. J. Cancer 2003, 104, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Maltais, R.; Trottier, A.; Roy, J.; Ayan, D.; Bertrand, N.; Poirier, D. Pharmacokinetic profile of PBRM in rodents, a first selective covalent inhibitor of 17β-HSD1 for breast cancer and endometriosis treatments. J. Steroid Biochem. Mol. Biol. 2018, 178, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Sasano, H.; Frost, A.R.; Saitoh, R.; Harada, N.; Poutanen, M.; Vihko, R.; Bulun, S.E.; Silverberg, S.G.; Nagura, H. Aromatase and 17β-hydroxysteroid dehydrogenase type 1 in human breast carcinoma. J. Clin. Endocrinol. Metab. 1996, 81, 4042–4046. [Google Scholar] [PubMed]
- Torn, S.; Nokelainen, P.; Kurkela, R.; Pulkka, A.; Menjivar, M.; Ghosh, S.; Coca-Prados, M.; Peltoketo, H.; Isomaa, V.; Vihko, P. Production, purification, and functional analysis of reconbinant human and mouse 17β-hydroxysteroid dehydrogenase type 7. Biochem. Biophys. Res. Commun. 2003, 305, 37–45. [Google Scholar] [CrossRef]
- Luu-The, V.; Tremblay, P.; Labrie, F. Characterization of type 12 17beta-hydroxystaeroid dehydrogenase, an isoform of type 13 17beta-hydroxysteroid dehydrogenase responsible for estradiol formation in women. Mol. Endocrinol. 2006, 20, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penning, T. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol. 2019, 489, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Miyagi, N.; Matsunaga, T.; Hara, A.; Ikari, A. Human dehydrogenase/reductase (SDR family) member 11 is a novel type of 17β-hydroxysteroid dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 472, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Maltais, R.; Poirier, D. Development of a gram-scale synthesis of PBRM, an irreversible inhibitor of 17β-hydroxysteroid dehydrogenase type 1. Org. Process Res. Dev. 2019, 23, 2323–2335. [Google Scholar] [CrossRef]
- Caron, C.; Turcotte, V.; Guillemette, C. A chromatography/tandem mass spectroscopy method for the simultaneous profiling of ten endogenous steroids, including progesterone, adrenal precursors, androgens and estrogens, using low serum volume. Steroids 2015, 104, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Lefebvre, J.; Maltais, R.; Poirier, D. Inhibition of dehydroepiandrosterone sulfate action in androgen-sensitive tissues by EM-1913, an inhibitor of steroid sulfatase. Mol. Cell. Endocrinol. 2013, 276, 148–155. [Google Scholar] [CrossRef]
- Kramer, C.Y. Extension of multiple range tests to group with unique numbers of replications. Biometrics 1956, 12, 307–310. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Steroids | LLOQ (ng/mL) | DHEA (ng/mL) | DHEA (%) | DHEA + PBRM (ng/mL) | DHEA + PBRM (%) | DHEA + LET (ng/mL) | DHEA + LET (%) | DHEA + PBRM + LET (ng/mL) | DHEA + PBRM + LET (%) |
|---|---|---|---|---|---|---|---|---|---|
| DHEA | 0.10 | 905 | 77.9 | 967 | 84.7 | 916 | 76.9 | 1051 | 85.0 |
| 5-diol | 0.05 | 244 | 21.0 | 159 | 13.9 | 261 | 21.9 | 169 | 13.7 |
| 4-dione | 0.05 | 12.1 | 1.04 | 15.7 | 1.4 | 14.0 | 1.2 | 16.4 | 1.32 |
| DHT | 0.01 | 0.34 | 0.03 | 0.23 | Tr | 0.23 | Tr | 0.24 | 0.02 |
| E2 | 0.005 | 0.18 | 0.01 | 0.20 | Tr | 0.22 | Tr | 0.22 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poirier, D.; Roy, J.; Maltais, R. A Targeted-Covalent Inhibitor of 17β-HSD1 Blocks Two Estrogen-Biosynthesis Pathways: In Vitro (Metabolism) and In Vivo (Xenograft) Studies in T-47D Breast Cancer Models. Cancers 2021, 13, 1841. https://doi.org/10.3390/cancers13081841
Poirier D, Roy J, Maltais R. A Targeted-Covalent Inhibitor of 17β-HSD1 Blocks Two Estrogen-Biosynthesis Pathways: In Vitro (Metabolism) and In Vivo (Xenograft) Studies in T-47D Breast Cancer Models. Cancers. 2021; 13(8):1841. https://doi.org/10.3390/cancers13081841
Chicago/Turabian StylePoirier, Donald, Jenny Roy, and René Maltais. 2021. "A Targeted-Covalent Inhibitor of 17β-HSD1 Blocks Two Estrogen-Biosynthesis Pathways: In Vitro (Metabolism) and In Vivo (Xenograft) Studies in T-47D Breast Cancer Models" Cancers 13, no. 8: 1841. https://doi.org/10.3390/cancers13081841