
An Alternative Pipeline for Glioblastoma Therapeutics: A Systematic Review of Drug Repurposing in Glioblastoma


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Discussion
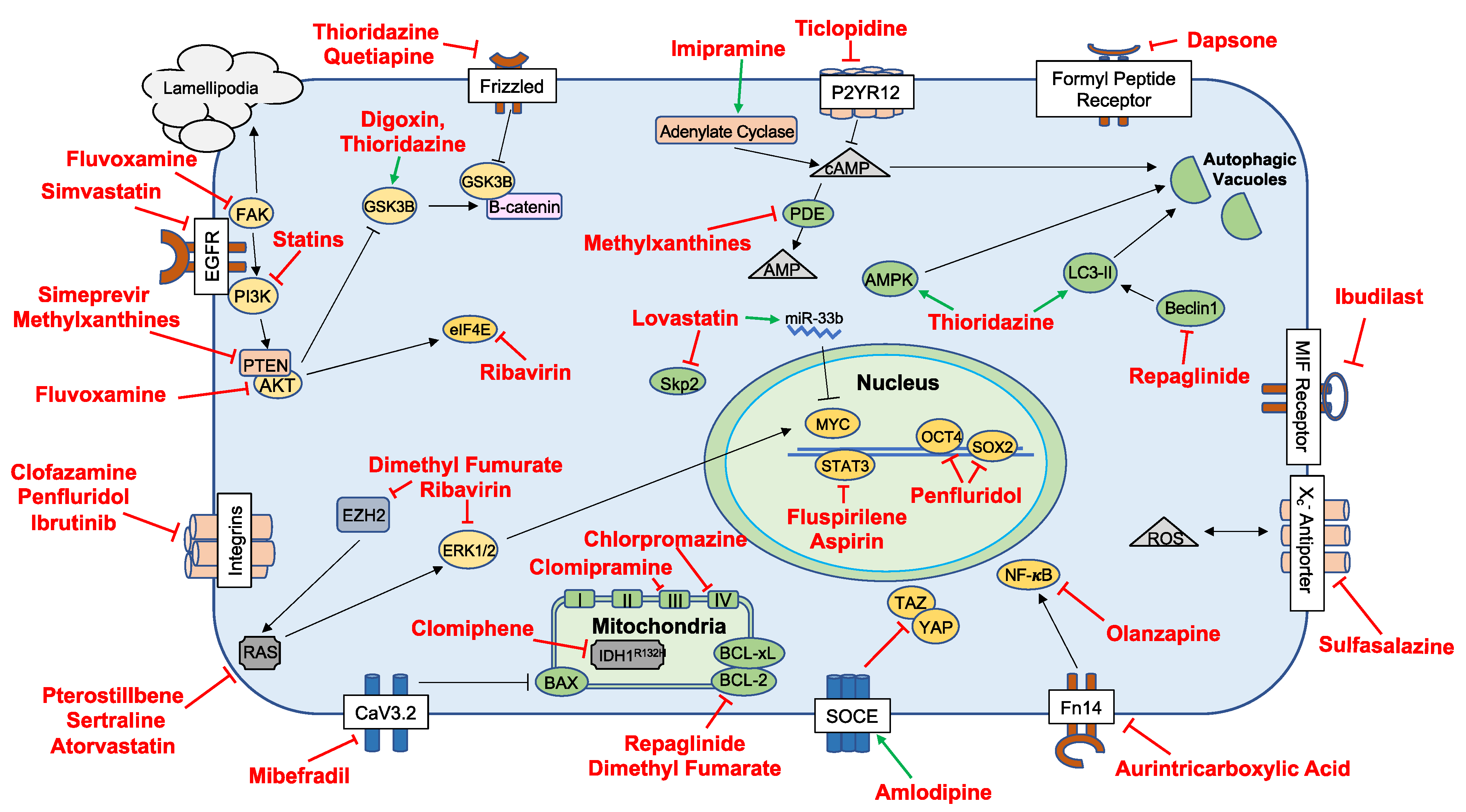
3.1. Repurposed Agents in Preclinical Study
3.1.1. Antiarrhythmics
3.1.2. Antibiotics-Tetracyclines, Macrolides, and Antimycobacterials
3.1.3. Antidiabetics
3.1.4. Antidepressants
3.1.5. Anti-Inflammatories
3.1.6. Immunosuppressants
3.1.7. Antihypertensives
3.1.8. Antipsychotics
3.1.9. Antivirals
3.1.10. Biologics and Small-Molecule Inhibitors
3.1.11. Disulfiram
3.1.12. Methylxanthines
3.1.13. Neurocognitive Agents
3.1.14. Statins
3.1.15. Other
3.1.16. Targeting the Tumor Microenvironment (TME)
3.1.17. Inhibition of Signaling Pathway Active in GBM
3.1.18. Targeting Glioma Stem Cells
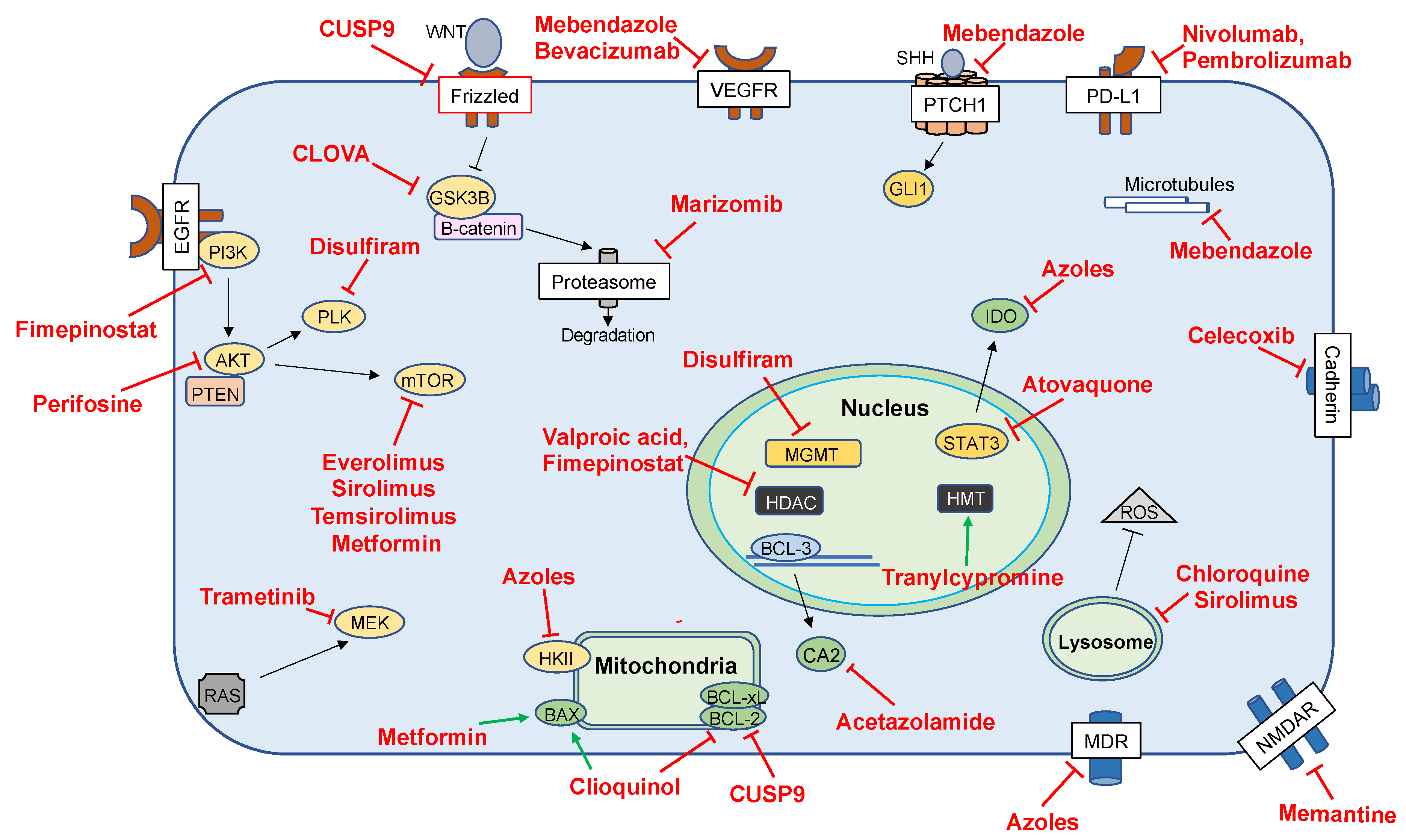
3.2. Repurposed Agents in Clinical Investigations
3.2.1. Antiepileptic Drugs (AEDs)
3.2.2. Disulfiram
3.2.3. Antifungals/Antimalarials
3.2.4. Antiparasitics
3.2.5. Antihypertensives
3.2.6. Anti-Inflammatories and Immunosuppressants
3.2.7. Antineoplastics
3.2.8. Carbonic-Anhydrase Inhibitors
3.2.9. Checkpoint Inhibitors
3.2.10. Diabetic Agents
3.2.11. Other Clinical Use of Small Molecules and Biologics
3.2.12. The Cocktails
3.2.13. Limitations
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Cantrell, J.N.; Waddle, M.R.; Rotman, M.; Peterson, J.L.; Ruiz-Garcia, H.; Heckman, M.G.; Quinones-Hinojosa, A.; Rosenfeld, S.S.; Brown, P.D.; Trifiletti, D.M. Progress Toward Long-Term Survivors of Glioblastoma. Mayo Clin. Proc. 2019, 94, 1278–1286. [Google Scholar] [CrossRef]
- Kast, R.E.; Boockvar, J.A.; Bruning, A.; Cappello, F.; Chang, W.W.; Cvek, B.; Dou, Q.P.; Duenas-Gonzalez, A.; Efferth, T.; Focosi, D.; et al. A conceptually new treatment approach for relapsed glioblastoma: Coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 2013, 4, 502–530. [Google Scholar] [CrossRef]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Patel, T.R.; Sirianni, R.W.; Strohbehn, G.; Zheng, M.Q.; Duong, N.; Schafbauer, T.; Huttner, A.J.; Huang, Y.; Carson, R.E.; et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc. Natl. Acad. Sci. USA 2013, 110, 11751–11756. [Google Scholar] [CrossRef] [PubMed]
- Determining Dose of Regadenoson Most Likely to Transiently Alter the Integrity of the Blood-Brain Barrier in Patients with High Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT03971734 (accessed on 1 February 2021).
- Intratumorally-Administered Topotecan Using Convection-Enhanced Delivery in Patients with Grade III/ IV Glioma. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03927274 (accessed on 1 February 2021).
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.D.; Mazandu, G.K.; Mulder, N.J. A systems-level analysis of drug-target-disease associations for drug repositioning. Brief. Funct. Genom. 2018, 17, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, Z.; Wang, B.; Xu, R. Towards precision medicine-based therapies for glioblastoma: Interrogating human disease genomics and mouse phenotypes. BMC Genom. 2016, 17 (Suppl. S7), 516. [Google Scholar] [CrossRef] [PubMed]
- Spina, R.; Voss, D.M.; Asnaghi, L.; Sloan, A.; Bar, E.E. Flow Cytometry-based Drug Screening System for the Identification of Small Molecules That Promote Cellular Differentiation of Glioblastoma Stem Cells. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Yang, E.H.; Shah, S.; Criley, J.M. Digitalis toxicity: A fading but crucial complication to recognize. Am. J. Med. 2012, 125, 337–343. [Google Scholar] [CrossRef]
- Berges, R.; Denicolai, E.; Tchoghandjian, A.; Baeza-Kallee, N.; Honore, S.; Figarella-Branger, D.; Braguer, D. Proscillaridin A exerts anti-tumor effects through GSK3beta activation and alteration of microtubule dynamics in glioblastoma. Cell Death Dis. 2018, 9, 984. [Google Scholar] [CrossRef]
- Tan, Q.; Yan, X.; Song, L.; Yi, H.; Li, P.; Sun, G.; Yu, D.; Li, L.; Zeng, Z.; Guo, Z. Induction of Mitochondrial Dysfunction and Oxidative Damage by Antibiotic Drug Doxycycline Enhances the Responsiveness of Glioblastoma to Chemotherapy. Med. Sci. Monit. 2017, 23, 4117–4125. [Google Scholar] [CrossRef]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Kast, R.E.; Siegelin, M.D.; Dwucet, A.; Schneider, E.; Westhoff, M.A.; Wirtz, C.R.; Chen, X.Y.; Halatsch, M.E.; Bolm, C. Anti-glioma Activity of Dapsone and Its Enhancement by Synthetic Chemical Modification. Neurochem. Res. 2017, 42, 3382–3389. [Google Scholar] [CrossRef]
- Mulkearns-Hubert, E.E.; Torre-Healy, L.A.; Silver, D.J.; Eurich, J.T.; Bayik, D.; Serbinowski, E.; Hitomi, M.; Zhou, J.; Przychodzen, B.; Zhang, R.; et al. Development of a Cx46 Targeting Strategy for Cancer Stem Cells. Cell Rep. 2019, 27, 1062–1072.e5. [Google Scholar] [CrossRef]
- Xiao, Z.X.; Chen, R.Q.; Hu, D.X.; Xie, X.Q.; Yu, S.B.; Chen, X.Q. Identification of repaglinide as a therapeutic drug for glioblastoma multiforme. Biochem. Biophys. Res. Commun. 2017, 488, 33–39. [Google Scholar] [CrossRef]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef]
- Keatley, K.; Stromei-Cleroux, S.; Wiltshire, T.; Rajala, N.; Burton, G.; Holt, W.V.; Littlewood, D.T.J.; Briscoe, A.G.; Jung, J.; Ashkan, K.; et al. Integrated Approach Reveals Role of Mitochondrial Germ-Line Mutation F18L in Respiratory Chain, Oxidative Alterations, Drug Sensitivity, and Patient Prognosis in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 3364. [Google Scholar] [CrossRef]
- Hayashi, K.; Michiue, H.; Yamada, H.; Takata, K.; Nakayama, H.; Wei, F.Y.; Fujimura, A.; Tazawa, H.; Asai, A.; Ogo, N.; et al. Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Sci. Rep. 2016, 6, 23372. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Baskaran, S.; Johansson, P.; Padhan, N.; Matuszewski, D.; Green, L.C.; Elfineh, L.; Wee, S.; Haggblad, M.; Martens, U.; et al. Case-specific potentiation of glioblastoma drugs by pterostilbene. Oncotarget 2016, 7, 73200–73215. [Google Scholar] [CrossRef]
- Mihajluk, K.; Simms, C.; Reay, M.; Madureira, P.A.; Howarth, A.; Murray, P.; Nasser, S.; Duckworth, C.A.; Pritchard, D.M.; Pilkington, G.J.; et al. IP1867B suppresses the insulin-like growth factor 1 receptor (IGF1R) ablating epidermal growth factor receptor inhibitor resistance in adult high grade gliomas. Cancer Lett. 2019, 458, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Takabe, H.; Warnken, Z.N.; Zhang, Y.; Davis, D.A.; Smyth, H.D.C.; Kuhn, J.G.; Weitman, S.; Williams Iii, R.O. A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component with a Polymer Carrier. Pharmaceutics 2018, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Assad Kahn, S.; Costa, S.L.; Gholamin, S.; Nitta, R.T.; Dubois, L.G.; Feve, M.; Zeniou, M.; Coelho, P.L.; El-Habr, E.; Cadusseau, J.; et al. The anti-hypertensive drug prazosin inhibits glioblastoma growth via the PKCdelta-dependent inhibition of the AKT pathway. EMBO Mol. Med. 2016, 8, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, Y.; Chen, Y.; Luo, Z.; Zhang, Z.; Sun, R.; Wan, Z.; Sun, J.; Lu, B.; Zhang, L.; et al. A novel immunochemotherapy based on targeting of cyclooxygenase and induction of immunogenic cell death. Biomaterials 2021, 270, 120708. [Google Scholar] [CrossRef] [PubMed]
- Assefnia, S.; Dakshanamurthy, S.; Guidry Auvil, J.M.; Hampel, C.; Anastasiadis, P.Z.; Kallakury, B.; Uren, A.; Foley, D.W.; Brown, M.L.; Shapiro, L.; et al. Cadherin-11 in poor prognosis malignancies and rheumatoid arthritis: Common target, common therapies. Oncotarget 2014, 5, 1458–1474. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Geeviman, K.; Ramulu, C.; Babu, P.P. The nonsteroidal anti-inflammatory drug celecoxib suppresses the growth and induces apoptosis of human glioblastoma cells via the NF-κB pathway. J. Neurooncol. 2012, 106, 99–109. [Google Scholar] [CrossRef]
- Shishodia, S.; Koul, D.; Aggarwal, B.B. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-kappa B activation through inhibition of activation of I kappa B alpha kinase and Akt in human non-small cell lung carcinoma: Correlation with suppression of COX-2 synthesis. J. Immunol. 2004, 173, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast sensitizes glioblastoma to temozolomide by targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 9, 2905. [Google Scholar] [CrossRef]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Forde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.H.; Wang, J.; et al. Drug repurposing: Sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Mattoo, A.R.; Joun, A.; Jessup, J.M. Repurposing of mTOR Complex Inhibitors Attenuates MCL-1 and Sensitizes to PARP Inhibition. Mol. Cancer Res. 2019, 17, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.P.C.; Kuo, J.S.; Chiang, H.C.; Wang, H.E.; Wang, Y.S.; Huang, C.C.; Huang, Y.C.; Chi, M.S.; Mehta, M.P.; Chi, K.H. Temozolomide, sirolimus and chloroquine is a new therapeutic combination that synergizes to disrupt lysosomal function and cholesterol homeostasis in GBM cells. Oncotarget 2018, 9, 6883–6896. [Google Scholar] [CrossRef]
- Perifosine and Torisel (Temsirolimus) for Recurrent/Progressive Malignant Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT02238496 (accessed on 1 February 2021).
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C.; et al. Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, R. Drug repurposing for glioblastoma based on molecular subtypes. J. Biomed. Inform. 2016, 64, 131–138. [Google Scholar] [CrossRef]
- Lee, H.; Kang, S.; Kim, W. Drug Repositioning for Cancer Therapy Based on Large-Scale Drug-Induced Transcriptional Signatures. PLoS ONE 2016, 11, e0150460. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; Liang, Y.H.; Kuo, Y.L.; Chuu, C.P.; Lin, C.Y.; Lee, M.H.; Wu, A.T.; Yeh, C.T.; Chen, E.I.; Whang-Peng, J.; et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015, 6, e1753. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.W.; Ko, H.J.; Chou, C.H.; Cheng, T.S.; Cheng, H.W.; Liang, Y.H.; Lai, Y.L.; Lin, C.Y.; Wang, C.; Loh, J.K.; et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/beta-Catenin Signaling Pathway in Glioma Cells. Int. J. Mol. Sci. 2019, 20, 473. [Google Scholar] [CrossRef]
- Johannessen, T.C.; Hasan-Olive, M.M.; Zhu, H.; Denisova, O.; Grudic, A.; Latif, M.A.; Saed, H.; Varughese, J.K.; Rosland, G.V.; Yang, N.; et al. Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. Int. J. Cancer 2019, 144, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Omoruyi, S.I.; Ekpo, O.E.; Semenya, D.M.; Jardine, A.; Prince, S. Exploitation of a novel phenothiazine derivative for its anti-cancer activities in malignant glioblastoma. Apoptosis 2020, 25, 261–274. [Google Scholar] [CrossRef]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.E.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 151, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Furuta, T.; Sabit, H.; Kitabayashi, T.; Jiapaer, S.; Kobayashi, M.; Ino, Y.; Todo, T.; Teng, L.; Hirao, A.; et al. Identification of antipsychotic drug fluspirilene as a potential anti-glioma stem cell drug. Oncotarget 2017, 8, 111728–111741. [Google Scholar] [CrossRef]
- Kim, H.; Chong, K.; Ryu, B.K.; Park, K.J.; Yu, M.O.; Lee, J.; Chung, S.; Choi, S.; Park, M.J.; Chung, Y.G.; et al. Repurposing Penfluridol in Combination with Temozolomide for the Treatment of Glioblastoma. Cancers 2019, 11, 1310. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Kast, R.E.; Westhoff, M.A.; Dwucet, A.; Welscher, N.; Nonnenmacher, L.; Hlavac, M.; Siegelin, M.D.; Wirtz, C.R.; Debatin, K.M.; et al. Olanzapine inhibits proliferation, migration and anchorage-independent growth in human glioblastoma cell lines and enhances temozolomide’s antiproliferative effect. J. Neurooncol. 2015, 122, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, N.; Li, H.; Liu, S.; Chen, X.; Yu, S.; Wu, N.; Bian, X.W.; Shen, H.Y.; Li, C.; et al. Promoting oligodendroglial-oriented differentiation of glioma stem cell: A repurposing of quetiapine for the treatment of malignant glioma. Oncotarget 2017, 8, 37511–37524. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamamoto, M.; Togashi, K.; Sanomachi, T.; Sugai, A.; Seino, S.; Yoshioka, T.; Kitanaka, C.; Okada, M. In vitro and in vivo anti-tumor effects of brexpiprazole, a newly-developed serotonin-dopamine activity modulator with an improved safety profile. Oncotarget 2019, 10, 3547–3558. [Google Scholar] [CrossRef]
- Kwon, J.; Kim, D.H.; Park, J.M.; Park, Y.H.; Hwang, Y.H.; Wu, H.G.; Shin, K.H.; Kim, I.A. Targeting Phosphatidylinositol 4-Kinase IIIalpha for Radiosensitization: A Potential Model of Drug Repositioning Using an Anti-Hepatitis C Viral Agent. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 867–876. [Google Scholar] [CrossRef]
- Volpin, F.; Casaos, J.; Sesen, J.; Mangraviti, A.; Choi, J.; Gorelick, N.; Frikeche, J.; Lott, T.; Felder, R.; Scotland, S.J.; et al. Use of an anti-viral drug, Ribavirin, as an anti-glioblastoma therapeutic. Oncogene 2017, 36, 3037–3047. [Google Scholar] [CrossRef]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Pandey, V.; Ranjan, N.; Narne, P.; Babu, P.P. Roscovitine effectively enhances antitumor activity of temozolomide in vitro and in vivo mediated by increased autophagy and Caspase-3 dependent apoptosis. Sci. Rep. 2019, 9, 5012. [Google Scholar] [CrossRef]
- Study of Binimetinib with Encorafenib in Adults with Recurrent BRAF V600-Mutated HGG. Available online: https://clinicaltrials.gov/ct2/show/NCT03973918 (accessed on 1 February 2021).
- Triscott, J.; Lee, C.; Hu, K.; Fotovati, A.; Berns, R.; Pambid, M.; Luk, M.; Kast, R.E.; Kong, E.; Toyota, E.; et al. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget 2012, 3, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Zhang, R.; Ali-Osman, F.; Bobustuc, G.C.; Srivenugopal, K.S. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2014, 35, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.; Wells, J.C.; Grinshtein, N.; King, J.C.; Hao, X.; Dang, N.H.; Wang, X.; Aman, A.; Uehling, D.; Datti, A.; et al. Disulfiram when Combined with Copper Enhances the Therapeutic Effects of Temozolomide for the Treatment of Glioblastoma. Clin. Cancer Res. 2016, 22, 3860–3875. [Google Scholar] [CrossRef] [PubMed]
- Madala, H.R.; Punganuru, S.R.; Ali-Osman, F.; Zhang, R.; Srivenugopal, K.S. Brain- and brain tumor-penetrating disulfiram nanoparticles: Sequence of cytotoxic events and efficacy in human glioma cell lines and intracranial xenografts. Oncotarget 2018, 9, 3459–3482. [Google Scholar] [CrossRef]
- Perez-Perez, D.; Reyes-Vidal, I.; Chavez-Cortez, E.G.; Sotelo, J.; Magana-Maldonado, R. Methylxanthines: Potential Therapeutic Agents for Glioblastoma. Pharmaceuticals 2019, 12, 130. [Google Scholar] [CrossRef]
- Sachkova, A.; Sperling, S.; Mielke, D.; Schatlo, B.; Rohde, V.; Ninkovic, M. Combined Applications of Repurposed Drugs and Their Detrimental Effects on Glioblastoma Cells. Anticancer Res. 2019, 39, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Serrato, A.; Saunders, J.T.; Holmes, B.; Nishimura, R.N.; Lichtenstein, A.; Gera, J. Repurposing Potential of Riluzole as an ITAF Inhibitor in mTOR Therapy Resistant Glioblastoma. Int. J. Mol. Sci. 2020, 21, 344. [Google Scholar] [CrossRef]
- Griffin, M.; Khan, R.; Basu, S.; Smith, S. Ion Channels as Therapeutic Targets in High Grade Gliomas. Cancers 2020, 12, 3068. [Google Scholar] [CrossRef]
- Booth, L.; Malkin, M.; Dent, P. Repurposing Tecfidera for cancer. Aging 2016, 8, 1289–1290. [Google Scholar] [CrossRef][Green Version]
- Damiani, E.; Yuecel, R.; Wallace, H.M. Repurposing of idebenone as a potential anti-cancer agent. Biochem. J. 2019, 476, 245–259. [Google Scholar] [CrossRef]
- Wu, J.; Su, H.K.; Yu, Z.H.; Xi, S.Y.; Guo, C.C.; Hu, Z.Y.; Qu, Y.; Cai, H.P.; Zhao, Y.Y.; Zhao, H.F.; et al. Skp2 modulates proliferation, senescence and tumorigenesis of glioma. Cancer Cell Int. 2020, 20, 71. [Google Scholar] [CrossRef]
- Takwi, A.A.; Li, Y.; Becker Buscaglia, L.E.; Zhang, J.; Choudhury, S.; Park, A.K.; Liu, M.; Young, K.H.; Park, W.Y.; Martin, R.C.; et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 2012, 4, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, X.; Wang, Q.; Li, W.; Yang, T. Inverse screening of Simvastatin kinase targets from glioblastoma druggable kinome. Comput. Biol. Chem. 2020, 86, 107243. [Google Scholar] [CrossRef]
- Peng, P.; Wei, W.; Long, C.; Li, J. Atorvastatin augments temozolomide’s efficacy in glioblastoma via prenylation-dependent inhibition of Ras signaling. Biochem. Biophys. Res. Commun. 2017, 489, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Dhruv, H.D.; Mathews, I.T.; Inge, L.J.; Tuncali, S.; Hartman, L.K.; Chow, D.; Millard, N.; Yin, H.H.; Kloss, J.; et al. Identification of aurintricarboxylic acid as a selective inhibitor of the TWEAK-Fn14 signaling pathway in glioblastoma cells. Oncotarget 2017, 8, 12234–12246. [Google Scholar] [CrossRef] [PubMed]
- Inada, M.; Shindo, M.; Kobayashi, K.; Sato, A.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.I. Anticancer effects of a non-narcotic opium alkaloid medicine, papaverine, in human glioblastoma cells. PLoS ONE 2019, 14, e0216358. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Sivakumar, K.C.; Mishra, R. Bacoside A Induces Tumor Cell Death in Human Glioblastoma Cell Lines through Catastrophic Macropinocytosis. Front. Mol. Neurosci. 2017, 10, 171. [Google Scholar] [CrossRef]
- Eales, K.L.; Wilkinson, E.A.; Cruickshank, G.; Tucker, J.H.R.; Tennant, D.A. Verteporfin selectively kills hypoxic glioma cells through iron-binding and increased production of reactive oxygen species. Sci. Rep. 2018, 8, 14358. [Google Scholar] [CrossRef]
- Zheng, M.; Sun, W.; Gao, S.; Luan, S.; Li, D.; Chen, R.; Zhang, Q.; Chen, L.; Huang, J.; Li, H. Structure based discovery of clomifene as a potent inhibitor of cancer-associated mutant IDH1. Oncotarget 2017, 8, 44255–44265. [Google Scholar] [CrossRef]
- Killick-Cole, C.L.; Singleton, W.G.B.; Bienemann, A.S.; Asby, D.J.; Wyatt, M.J.; Boulter, L.J.; Barua, N.U.; Gill, S.S. Repurposing the anti-epileptic drug sodium valproate as an adjuvant treatment for diffuse intrinsic pontine glioma. PLoS ONE 2017, 12, e0176855. [Google Scholar] [CrossRef]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.; Braga, C.; Santos, G.; Bronze, M.R.; Perry, M.J.; Moreira, R.; Brites, D.; Falcao, A.S. Targeting Gliomas: Can a New Alkylating Hybrid Compound Make a Difference? ACS Chem. Neurosci. 2017, 8, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Sorafenib Tosylate, Valproic Acid, and Sildenafil Citrate in Treating Patients with Recurrent High-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT01817751 (accessed on 1 February 2021).
- Valproic Acid and Radiation Followed by Maintenance Valproic Acid and Bevacizumab in Children with High Grade Gliomas or Diffuse Intrinsic Pontine Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT00879437 (accessed on 1 February 2021).
- Huang, J.; Campian, J.L.; Gujar, A.D.; Tran, D.D.; Lockhart, A.C.; DeWees, T.A.; Tsien, C.I.; Kim, A.H. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. J. Neurooncol. 2016, 128, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Jakola, A.S.; Werlenius, K.; Mudaisi, M.; Hylin, S.; Kinhult, S.; Bartek, J., Jr.; Salvesen, O.; Carlsen, S.M.; Strandeus, M.; Lindskog, M.; et al. Disulfiram repurposing combined with nutritional copper supplement as add-on to chemotherapy in recurrent glioblastoma (DIRECT): Study protocol for a randomized controlled trial. F1000Res 2018, 7, 1797. [Google Scholar] [CrossRef]
- Disulfiram in Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02678975 (accessed on 1 February 2021).
- Poser, S.W.; Otto, O.; Arps-Forker, C.; Ge, Y.; Herbig, M.; Andree, C.; Gruetzmann, K.; Adasme, M.F.; Stodolak, S.; Nikolakopoulou, P.; et al. Controlling distinct signaling states in cultured cancer cells provides a new platform for drug discovery. FASEB J. 2019, 33, 9235–9249. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef]
- Azoles Targeting Recurrent High Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT03763396 (accessed on 1 February 2021).
- Wehbe, M.; Malhotra, A.K.; Anantha, M.; Lo, C.; Dragowska, W.H.; Dos Santos, N.; Bally, M.B. Development of a copper-clioquinol formulation suitable for intravenous use. Drug Deliv. Transl. Res. 2018, 8, 239–251. [Google Scholar] [CrossRef]
- Shipman, L. Glioma: Repurposed drugs combined to amplify autophagy. Nat. Rev. Cancer 2015, 15, 636. [Google Scholar] [CrossRef]
- Hribar, K.C.; Wheeler, C.J.; Bazarov, A.; Varshneya, K.; Yamada, R.; Buckley, P.; Patil, C.G. A Simple Three-dimensional Hydrogel Platform Enables Ex Vivo Cell Culture of Patient and PDX Tumors for Assaying Their Response to Clinically Relevant Therapies. Mol. Cancer Ther. 2019, 18, 718–725. [Google Scholar] [CrossRef]
- A Trial of Dabrafenib, Trametinib and Hydroxychloroquine for Patients with Recurrent LGG or HGG with a BRAF Aberration. Available online: https://clinicaltrials.gov/ct2/show/NCT04201457 (accessed on 1 February 2021).
- Maraka, S.; Groves, M.D.; Penas-Prado, M. Reply to Unexpectedly low rates of neuropsychiatric adverse effects associated with mefloquine repurposed for the treatment of glioblastoma. Cancer 2019, 125, 1385–1386. [Google Scholar] [CrossRef]
- Nevin, R.L. Unexpectedly low rates of neuropsychiatric adverse effects associated with mefloquine repurposed for the treatment of glioblastoma. Cancer 2019, 125, 1384–1385. [Google Scholar] [CrossRef] [PubMed]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; O’Brien, B.J.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2019, 125, 424–433. [Google Scholar] [CrossRef]
- Bai, R.Y.; Staedtke, V.; Rudin, C.M.; Bunz, F.; Riggins, G.J. Effective treatment of diverse medulloblastoma models with mebendazole and its impact on tumor angiogenesis. Neuro Oncol. 2015, 17, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Bodhinayake, I.; Symons, M.; Boockvar, J.A. Repurposing mebendazole for the treatment of medulloblastoma. Neurosurgery 2015, 76, N15–N16. [Google Scholar] [CrossRef] [PubMed]
- De Witt, M.; Gamble, A.; Hanson, D.; Markowitz, D.; Powell, C.; Al Dimassi, S.; Atlas, M.; Boockvar, J.; Ruggieri, R.; Symons, M. Repurposing Mebendazole as a Replacement for Vincristine for the Treatment of Brain Tumors. Mol. Med. 2017, 23, 50–56. [Google Scholar] [CrossRef]
- Larsen, A.R.; Bai, R.Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Phase I Study of Mebendazole Therapy for Recurrent/Progressive Pediatric Brain Tumors. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02644291 (accessed on 1 February 2021).
- Happold, C.; Gorlia, T.; Nabors, L.B.; Erridge, S.C.; Reardon, D.A.; Hicking, C.; Picard, M.; Stupp, R.; Weller, M.; Group, E.B.T.; et al. Do statins, ACE inhibitors or sartans improve outcome in primary glioblastoma? J. Neurooncol. 2018, 138, 163–171. [Google Scholar] [CrossRef]
- Ursu, R.; Thomas, L.; Psimaras, D.; Chinot, O.; Le Rhun, E.; Ricard, D.; Charissoux, M.; Cuzzubbo, S.; Sejalon, F.; Quillien, V.; et al. Angiotensin II receptor blockers, steroids and radiotherapy in glioblastoma-a randomised multicentre trial (ASTER trial). An ANOCEF study. Eur. J. Cancer 2019, 109, 129–136. [Google Scholar] [CrossRef]
- Reardon, D.A.; Quinn, J.A.; Vredenburgh, J.; Rich, J.N.; Gururangan, S.; Badruddoja, M.; Herndon, J.E.; Dowell, J.M.; Friedman, A.H.; Friedman, H.S. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer 2005, 103, 329–338. [Google Scholar] [CrossRef]
- Kesari, S.; Schiff, D.; Henson, J.W.; Muzikansky, A.; Gigas, D.C.; Doherty, L.; Batchelor, T.T.; Longtine, J.A.; Ligon, K.L.; Weaver, S.; et al. Phase II study of temozolomide, thalidomide, and celecoxib for newly diagnosed glioblastoma in adults. Neuro Oncol. 2008, 10, 300–308. [Google Scholar] [CrossRef]
- Levin, V.A.; Giglio, P.; Puduvalli, V.K.; Jochec, J.; Groves, M.D.; Yung, W.K.; Hess, K. Combination chemotherapy with 13-cis-retinoic acid and celecoxib in the treatment of glioblastoma multiforme. J. Neurooncol. 2006, 78, 85–90. [Google Scholar] [CrossRef]
- A Study of Ribociclib and Everolimus Following Radiation Therapy in Children With Newly Diagnosed Non-biopsied Diffuse Pontine Gliomas (DIPG) and RB+ Biopsied DIPG and High Grade Gliomas (HGG). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03355794 (accessed on 1 February 2021).
- Phase I–II Everolimus and Sorafenib in Recurrent High-Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT01434602 (accessed on 1 February 2021).
- A Phase 0 II Study of Ribociclib (LEE011) in Combination With Everolimus in Preoperative Recurrent High-Grade Glioma Patients Scheduled for Resection. Available online: https://clinicaltrials.gov/ct2/show/NCT03834740 (accessed on 1 February 2021).
- Study of Dasatinib in Combination with Everolimus for Children and Young Adults with Gliomas Harboring PDGFR Alterations. Available online: https://clinicaltrials.gov/ct2/show/NCT03352427 (accessed on 1 February 2021).
- Ribociclib and Everolimus in Treating Children with Recurrent or Refractory Malignant Brain Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03387020 (accessed on 1 February 2021).
- Vorinostat and Temozolomide in Treating Patients with Malignant Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT00268385 (accessed on 1 February 2021).
- Pilot Study of Cabozantinib for Recurrent or Progressive High-Grade Glioma in Children. Available online: https://clinicaltrials.gov/ct2/show/NCT02885324 (accessed on 1 February 2021).
- Arsenic Trioxide, Temozolomide, and Radiation Therapy in Treating Patients with Malignant Glioma That Has Been Removed by Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT00275067 (accessed on 1 February 2021).
- Stage 1: Marizomib + Bevacizumab in WHO Gr IV GBM.; Stage 2: Marizomib Alone; Stage 3: Combination of Marizomib and Bevacizumab. Available online: https://clinicaltrials.gov/ct2/show/NCT02330562 (accessed on 1 February 2021).
- Study of Marizomib with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Brain Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02903069 (accessed on 1 February 2021).
- Fimepinostat in Treating Brain Tumors in Children and Young Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT03893487 (accessed on 1 February 2021).
- Wu, L.; Bernal, G.M.; Cahill, K.E.; Pytel, P.; Fitzpatrick, C.A.; Mashek, H.; Weichselbaum, R.R.; Yamini, B. BCL3 expression promotes resistance to alkylating chemotherapy in gliomas. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Das, A.; Banik, N.L.; Ray, S.K. Modulatory effects of acetazolomide and dexamethasone on temozolomide-mediated apoptosis in human glioblastoma T98G and U87MG cells. Cancer Investig. 2008, 26, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Study of Acetazolamide with Temozolomide in Adults with Newly Diagnosed or Recurrent Malignant Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT03011671 (accessed on 1 February 2021).
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Neoadjuvant Nivolumab in Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02550249 (accessed on 1 February 2021).
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- An Investigational Immuno-Therapy Study of Nivolumab Compared to Temozolomide, Each Given with Radiation Therapy, for Newly-Diagnosed Patients with Glioblastoma (GBM, a Malignant Brain Cancer). Available online: https://clinicaltrials.gov/ct2/show/NCT02617589 (accessed on 1 February 2021).
- An Investigational Immuno-Therapy Study of Temozolomide Plus Radiation Therapy with Nivolumab or Placebo, for Newly Diagnosed Patients with Glioblastoma (GBM, a Malignant Brain Cancer). Available online: https://clinicaltrials.gov/ct2/show/NCT02667587 (accessed on 1 February 2021).
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; Groot, J.F.D.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; Gaffey, S.C.; et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2018, 36, 2006. [Google Scholar] [CrossRef]
- Pembrolizumab in Treating Younger Patients with Recurrent, Progressive, or Refractory High-Grade Gliomas, Diffuse Intrinsic Pontine Gliomas, Hypermutated Brain Tumors, Ependymoma or Medulloblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02359565 (accessed on 1 February 2021).
- Hypofractionated Stereotactic Irradiation (HFSRT) with Pembrolizumab and Bevacizumab for Recurrent High Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT02313272 (accessed on 1 February 2021).
- Gritti, M.; Wurth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef]
- Rezaei, N.; Neshasteh-Riz, A.; Mazaheri, Z.; Koosha, F.; Hoormand, M. The Combination of Metformin and Disulfiram-Cu for Effective Radiosensitization on Glioblastoma Cells. Cell J. 2020, 22, 263–272. [Google Scholar] [CrossRef]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef]
- Seliger, C.; Luber, C.; Gerken, M.; Schaertl, J.; Proescholdt, M.; Riemenschneider, M.J.; Meier, C.R.; Bogdahn, U.; Leitzmann, M.F.; Klinkhammer-Schalke, M.; et al. Use of metformin and survival of patients with high-grade glioma. Int. J. Cancer 2019, 144, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P.; Group, E.B.T. Use of metformin and outcome of patients with newly diagnosed glioblastoma: Pooled analysis. Int. J. Cancer 2020, 146, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, M.W.A.; van Linde, M.E.; Kouwenhoven, M.; Bramer, J.A.M.; Bovee, J.; Mathot, R.A.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef]
- Temozolomide, Memantine Hydrochloride, Mefloquine, and Metformin Hydrochloride in Treating Patients with Glioblastoma Multiforme after Radiation Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT01430351 (accessed on 1 February 2021).
- Metformin and Chloroquine in IDH1/2-Mutated Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02496741 (accessed on 1 February 2021).
- Treatment of Recurrent Brain Tumors: Metabolic Manipulation Combined with Radiotherapy. Available online: https://clinicaltrials.gov/ct2/show/NCT02149459 (accessed on 1 February 2021).
- Bioavailability of Disulfiram and Metformin in Glioblastomas. Available online: https://clinicaltrials.gov/ct2/show/NCT03151772 (accessed on 1 February 2021).
- Metformin, Neo-Adjuvant Temozolomide and Hypo-Accelerated Radiotherapy Followed by Adjuvant TMZ in Patients with GBM. Available online: https://clinicaltrials.gov/ct2/show/NCT02780024 (accessed on 1 February 2021).
- Study on Low Dose Temozolomide Plus Metformin or Placebo in Patient with Recurrent or Refractory Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03243851 (accessed on 1 February 2021).
- Miller, S.E.; Tummers, W.S.; Teraphongphom, N.; van den Berg, N.S.; Hasan, A.; Ertsey, R.D.; Nagpal, S.; Recht, L.D.; Plowey, E.D.; Vogel, H.; et al. First-in-human intraoperative near-infrared fluorescence imaging of glioblastoma using cetuximab-IRDye800. J. Neurooncol. 2018, 139, 135–143. [Google Scholar] [CrossRef]
- A Study of the Specificity and Sensitivity of 5- Aminolevulinic Acid (ALA) Fluorescence in Malignant Brain Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT01128218 (accessed on 1 February 2021).
- ALA-induced PpIX Fluorescence during Brain Tumor Resection. Available online: https://clinicaltrials.gov/ct2/show/NCT02191488 (accessed on 1 February 2021).
- Diagnostic Performance of Fluorescein as an Intraoperative Brain Tumor Biomarker. Available online: https://clinicaltrials.gov/ct2/show/NCT02691923 (accessed on 1 February 2021).
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; Group, A.-G.S. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Furuta, T.; Sabit, H.; Dong, Y.; Miyashita, K.; Kinoshita, M.; Uchiyama, N.; Hayashi, Y.; Hayashi, Y.; Minamoto, T.; Nakada, M. Biological basis and clinical study of glycogen synthase kinase- 3beta-targeted therapy by drug repositioning for glioblastoma. Oncotarget 2017, 8, 22811–22824. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Kast, R.E.; Dwucet, A.; Hlavac, M.; Heiland, T.; Westhoff, M.A.; Debatin, K.M.; Wirtz, C.R.; Siegelin, M.D.; Karpel-Massler, G. Bcl-2/Bcl-xL inhibition predominantly synergistically enhances the anti-neoplastic activity of a low-dose CUSP9 repurposed drug regime against glioblastoma. Br. J. Pharmacol. 2019, 176, 3681–3694. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Skaga, I.O.; Grieg, Z.; Sandberg, C.J.; Langmoen, I.A.; Vik-Mo, E.O. The efficacy of a coordinated pharmacological blockade in glioblastoma stem cells with nine repurposed drugs using the CUSP9 strategy. J. Cancer Res. Clin. Oncol. 2019, 145, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef]
- A Proof-of-Concept Clinical Trial Assessing the Safety of the Coordinated Undermining of Survival Paths by 9 Repurposed Drugs Combined with Metronomic Temozolomide (CUSP9v3 Treatment Protocol) for Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02770378 (accessed on 1 February 2021).
- Verschuur, A.; Heng-Maillard, M.A.; Dory-Lautrec, P.; Truillet, R.; Jouve, E.; Chastagner, P.; Leblond, P.; Aerts, I.; Honore, S.; Entz-Werle, N.; et al. Metronomic Four-Drug Regimen Has Anti-tumor Activity in Pediatric Low-Grade Glioma; The Results of a Phase II Clinical Trial. Front. Pharmacol. 2018, 9, 00950. [Google Scholar] [CrossRef]
- Huang, L.; Garrett Injac, S.; Cui, K.; Braun, F.; Lin, Q.; Du, Y.; Zhang, H.; Kogiso, M.; Lindsay, H.; Zhao, S.; et al. Systems biology-based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Lirk, P.; Tan, C.H.; Seymour, R.A. An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin. Med. Res. 2007, 5, 19–34. [Google Scholar] [CrossRef]
- Galvao, R.P.; Zong, H. Inflammation and Gliomagenesis: Bi-Directional Communication at Early and Late Stages of Tumor Progression. Curr. Pathobiol. Rep. 2013, 1, 19–28. [Google Scholar] [CrossRef]
- Goglia, A.G.; Delsite, R.; Luz, A.N.; Shahbazian, D.; Salem, A.F.; Sundaram, R.K.; Chiaravalli, J.; Hendrikx, P.J.; Wilshire, J.A.; Jasin, M.; et al. Identification of novel radiosensitizers in a high-throughput, cell-based screen for DSB repair inhibitors. Mol. Cancer Ther. 2015, 14, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.P. The withdrawal of mibefradil (Posicor). Advers. Drug React. Toxicol. Rev. 1998, 17, 59–60. [Google Scholar]
- Vellichirammal, N.N.; Albahrani, A.; Banwait, J.K.; Mishra, N.K.; Li, Y.; Roychoudhury, S.; Kling, M.J.; Mirza, S.; Bhakat, K.K.; Band, V.; et al. Pan-Cancer Analysis Reveals the Diverse Landscape of Novel Sense and Antisense Fusion Transcripts. Mol. Ther. Nucleic Acids 2020, 19, 1379–1398. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Chatziathanasiadou, M.V.; Stylos, E.K.; Giannopoulou, E.; Spyridaki, M.H.; Briasoulis, E.; Kalofonos, H.P.; Crook, T.; Syed, N.; Sivolapenko, G.B.; Tzakos, A.G. Development of a validated LC-MS/MS method for the in vitro and in vivo quantitation of sunitinib in glioblastoma cells and cancer patients. J. Pharm. Biomed. Anal. 2019, 164, 690–697. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Seliger, C.; Schaertl, J.; Gerken, M.; Luber, C.; Proescholdt, M.; Riemenschneider, M.J.; Leitzmann, M.F.; Hau, P.; Klinkhammer-Schalke, M. Use of statins or NSAIDs and survival of patients with high-grade glioma. PLoS ONE 2018, 13, e0207858. [Google Scholar] [CrossRef]
- Pointer, K.B.; Clark, P.A.; Eliceiri, K.W.; Salamat, M.S.; Robertson, G.A.; Kuo, J.S. Administration of Non-Torsadogenic human Ether-a-go-go-Related Gene Inhibitors Is Associated with Better Survival for High hERG-Expressing Glioblastoma Patients. Clin. Cancer Res. 2017, 23, 73–80. [Google Scholar] [CrossRef]
- Brown, B.M.; Pressley, B.; Wulff, H. KCa3.1 Channel Modulators as Potential Therapeutic Compounds for Glioblastoma. Curr. Neuropharmacol. 2018, 16, 618–626. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Tran, A.; Nghiemphu, P.L.; Pope, W.B.; Solis, O.E.; Selch, M.; Filka, E.; Yong, W.H.; Mischel, P.S.; Liau, L.M.; et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J. Clin. Oncol. 2011, 29, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Vredenburgh, J.J.; Desjardins, A.; Reardon, D.A.; Peters, K.B.; Herndon, J.E.; Marcello, J.; Kirkpatrick, J.P.; Sampson, J.H.; Bailey, L.; Threatt, S.; et al. The addition of bevacizumab to standard radiation therapy and temozolomide followed by bevacizumab, temozolomide, and irinotecan for newly diagnosed glioblastoma. Clin. Cancer Res. 2011, 17, 4119–4124. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Matteoni, S.; Matarrese, P.; Ascione, B.; Buccarelli, M.; Ricci-Vitiani, L.; Pallini, R.; Villani, V.; Pace, A.; Paggi, M.G.; Abbruzzese, C. Anticancer Properties of the Antipsychotic Drug Chlorpromazine and Its Synergism with Temozolomide in Restraining Human Glioblastoma Proliferation. Front. Oncol. 2021, 11, 635472. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Dong, X.; Li, H.; Wang, H.; Jiang, Q.; Liu, L.; Wang, L.; Dong, J. Nicardipine sensitizes temozolomide by inhibiting autophagy and promoting cell apoptosis in glioma stem cells. Aging 2021, 13, 6820–6831. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Sears, T.; Cortopassi, G.; Woolard, K.; Angelastro, J.M. Repurposing FDA approved drugs inhibiting mitochondrial function for targeting glioma-stem like cells. Biomed. Pharmacother. 2021, 133, 111058. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Okada, M.; Yoshioka, T.; Kitanaka, C. Doxazosin, a Classic Alpha 1-Adrenoceptor Antagonist, Overcomes Osimertinib Resistance in Cancer Cells via the Upregulation of Autophagy as Drug Repurposing. Biomedicines 2020, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Toscano, A.; Khan, D.; Nickel, A.C.; Hewera, M.; Kamp, M.A.; Fischer, I.; Steiger, H.J.; Zhang, W.; Muhammad, S.; Hänggi, D.; et al. Robot technology identifies a Parkinsonian therapeutics repurpose to target stem cells of glioblastoma. CNS Oncol. 2020, 9, CNS58. [Google Scholar] [CrossRef]
- Knudsen-Baas, K.M.; Engeland, A.; Gilhus, N.E.; Storstein, A.M.; Owe, J.F. Does the choice of antiepileptic drug affect survival in glioblastoma patients? J. Neurooncol. 2016, 129, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Toovey, S. Mefloquine neurotoxicity: A literature review. Travel Med. Infect. Dis. 2009, 7, 2–6. [Google Scholar] [CrossRef]
- Brazil, L.; Swampillai, A.L.; Mak, K.M.; Edwards, D.; Mesiri, P.; Clifton-Hadley, L.; Shaffer, R.; Lewis, J.; Watts, C.; Jeffries, S.; et al. Hydroxychloroquine and short-course radiotherapy in elderly patients with newly diagnosed high-grade glioma: A randomized phase II trial. Neurooncol. Adv. 2020, 2, vdaa046. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Hejazi, S.; Hiddingh, L.; Carvalho, L.; de Gooijer, M.C.; Wakimoto, H.; Barazas, M.; Tannous, M.; Chi, A.S.; Noske, D.P.; et al. Recycling drug screen repurposes hydroxyurea as a sensitizer of glioblastomas to temozolomide targeting de novo DNA synthesis, irrespective of molecular subtype. Neuro Oncol. 2018, 20, 642–654. [Google Scholar] [CrossRef]
- Yamashita, A.S.; da Costa Rosa, M.; Borodovsky, A.; Festuccia, W.T.; Chan, T.; Riggins, G.J. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro Oncol. 2019, 21, 189–200. [Google Scholar] [CrossRef]
- Taylor, J.T.; Ellison, S.; Pandele, A.; Wood, S.; Nathan, E.; Forte, G.; Parker, H.; Zindy, E.; Elvin, M.; Dickson, A.; et al. Actinomycin D Downregulates Sox2 and Improves Survival in Preclinical Models of Recurrent Glioblastoma. Neuro Oncol. 2020. [Google Scholar] [CrossRef]
- Wehbe, M.; Anantha, M.; Shi, M.; Leung, A.W.; Dragowska, W.H.; Sanche, L.; Bally, M.B. Development and optimization of an injectable formulation of copper diethyldithiocarbamate, an active anticancer agent. Int. J. Nanomed. 2017, 12, 4129–4146. [Google Scholar] [CrossRef]
- Takahashi, M.; Miki, S.; Fujimoto, K.; Fukuoka, K.; Matsushita, Y.; Maida, Y.; Yasukawa, M.; Hayashi, M.; Shinkyo, R.; Kikuchi, K.; et al. Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 2019, 110, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, C.A.; Nguyen, H.P.T.; Morokoff, A.P.; Luwor, R.B.; Paradiso, L.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S. Inhibition of Radiation and Temozolomide-Induced Invadopodia Activity in Glioma Cells Using FDA-Approved Drugs. Transl. Oncol. 2018, 11, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Afjei, R.; Sekar, T.V.; Babikir, H.A.; Massoud, T.F. A protein folding molecular imaging biosensor monitors the effects of drugs that restore mutant p53 structure and its downstream function in glioblastoma cells. Oncotarget 2018, 9, 21495–21511. [Google Scholar] [CrossRef][Green Version]
- Dave, N.; Chow, L.M.; Gudelsky, G.A.; LaSance, K.; Qi, X.; Desai, P.B. Preclinical pharmacological evaluation of letrozole as a novel treatment for gliomas. Mol. Cancer Ther. 2015, 14, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Tivnan, A.; Heilinger, T.; Ramsey, J.M.; O’Connor, G.; Pokorny, J.L.; Sarkaria, J.N.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Kim, E.L.; et al. Anti-GD2-ch14.18/CHO coated nanoparticles mediate glioblastoma (GBM)-specific delivery of the aromatase inhibitor, Letrozole, reducing proliferation, migration and chemoresistance in patient-derived GBM tumor cells. Oncotarget 2017, 8, 16605–16620. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [Google Scholar] [CrossRef] [PubMed]
- A Pilot Surgical Trial to Evaluate Early Immunologic Pharmacodynamic Parameters for the PD-1 Checkpoint Inhibitor, Pembrolizumab (MK-3475), in Patients with Surgically Accessible Recurrent/Progressive Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02852655 (accessed on 1 February 2021).
- Efficacy of Nivolumab for Recurrent IDH Mutated High-Grade Gliomas. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03925246 (accessed on 1 February 2021).
- Kast, R.E.; Skuli, N.; Karpel-Massler, G.; Frosina, G.; Ryken, T.; Halatsch, M.E. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: The EIS regimen. Oncotarget 2017, 8, 60727–60749. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Dietrich, P.Y.; Stupp, R.; Linette, G.P.; Posey, A.D.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [PubMed]
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug Class | Drugs | Targets | Reference(s) |
|---|---|---|---|
| Antiarrhythmics | Digoxin | Na+/K+ ATPase, AKT | [19] |
| Proscillaridin A | GSK3β | [20] | |
| Antibiotics | Tetracyclines | Mitochondria | [21,22] |
| Dapsone | FPR, IL-8, Leukotriene-B4 | [23] | |
| Clofazimine | Cx46 | [24] | |
| Antidiabetics | Repaglinide | BCL-2, PD-L1, Beclin 1 | [25] |
| Antidepressants | Imipramine | ATG7 | [26] |
| Clomipramine | Complex III Cytochrome B | [27] | |
| Fluvoxamine | AKT/mTOR | [28] | |
| Sertraline | MAPK | [29] | |
| Anti-inflammatories | IP187B/Aspirin | STAT3, NF-κB, IGFR, PD-1 | [28,30,31,32,33] |
| Celecoxib | COX-2, NF-κB | [34,35,36] | |
| Ibudilast | MIF | [37] | |
| Sulfasalazine | System X(c)(-) Antiporter | [38,39] | |
| Immunosuppressants | Everolimus | mTOR, MCL-1 | [40,41,42] |
| Sirolimus | |||
| Temsirolimus | |||
| Antihypertensives | Mibefradil | NHEJ, Cav3.2 | [43] |
| Prazosin | AKT | [32] | |
| Amlodipine | PKD, Caspase 3 | [44] | |
| Pentoxifylline | NA | [45] | |
| Antipsychotics | Thioridazine | AMPK, MAP1/LC3-II, WNT | [46,47,48] |
| DS00329 | Cyclin A, Cyclin B, Cyclin D1 | [49] | |
| Chlorpromazine | CcO Complex IV | [50,51] | |
| Fluspirilene | STAT3 | [52] | |
| Penfluridol | SOX2, OCT4, uPAR | [53] | |
| Olanzapine | AMPK | [54] | |
| Quetiapine | WNT | [55] | |
| Brexpiprazole | Survivin | [56] | |
| Antivirals | Simeprevir | PI4K | [57] |
| Ribavirin | EZH2, ERK | [58] | |
| Biologics and Small-Molecule Inhibitors | AS602801 | JNK | [59] |
| CEP-1347 | [60] | ||
| LY294002 | PI3K | [61] | |
| PX-886 | [61] | ||
| Ibrutinib | TK, BMX-STA3 | [62] | |
| Roscovitin | CDK | [63] | |
| Binimetinib | MEK | [64] | |
| Encorafenib | BRAF | [64] | |
| Disulfiram | Disulfiram | PLK1, Ubiquitin–Proteasome Pathway, AIF | [65,66,67,68] |
| Methylxanthines | Theophylline | PDE | |
| Theobromine | [69] | ||
| Caffeine | |||
| Neurocognitive | Riluzole | Na+ Transporter, ITAF hnRNP A1, HIF1A, AKT | [70,71,72] |
| Dimethyl fumarate | ERK1/2, AKT | [73] | |
| Idebenone | p21 | [74] | |
| Statins | Lovastatin | c-Myc, SKP2 | [75,76] |
| Simvastatin | EGFR, FGFR, c-SRC | [77] | |
| Atorvastatin | RAS | [78] | |
| Other | Aurintricarboxylic acid | NF-κB | [79] |
| Papaverine | HMGB1/RAGE | [80] | |
| Bacoside A | CAMKIIA | [81] | |
| Verteporfin | YAP, HIF1A | [82] | |
| Clomiphene | IDH1 | [83] |
| Drug Class | Drugs | Targets | Clinical Trial Stage | References |
|---|---|---|---|---|
| Antiepileptic Drugs | Valproic Acid | HDAC | Phase II | [70,84,85,86,87,88] |
| Disulfiram | Disulfiram | PLK1, Ubiquitin–Proteasome Pathway, AIF | Phase II/III | [89,90,91] |
| Antifungals | Azoles | Hexokinase II | Phase I | [92,93,94] |
| Clioquinol | BAX, BCL-2 | Phase I | [92,95] | |
| Antimalarials | Atovaquone | STAT3 | Preclinical | [31] |
| Chloroquine | Unclear | Preclinical | [26,41,86,96,97] | |
| Hydroxychloroquine | LC3-II | Phase I/II | [98] | |
| Mefloquine | NMDA | Phase I/II | [99,100,101] | |
| Antiparasitics | Mebendazole | Microtubules, VEGF | Phase I | [102,103,104,105,106] |
| Antihypertensives | ARBs, ACEis | Unclear | Retrospective | [32,44,45,107,108] |
| Anti-inflammatories | Celecoxib | COX-2 | Phase I–II | [109,110,111] |
| Immunosuppressants | Temsirolimus | mTOR, MCL-1 | Phase I | [42] |
| Everolimus | Phase I/II | [112,113,114,115,116] | ||
| Antineoplastics | Vorinostat | HDAC | Phase I/II | [117] |
| Cabozantinib | TK | Phase II | [118] | |
| Arsenic Trioxide | Cytochrome C | Phase I/II | [119] | |
| Marizomib | Proteasome | Phase I | [120,121] | |
| Fimepinostat | PI3K, HDAC | Phase I | [122] | |
| Carbonic-Anhydrase Inhibitors | Acetazolamide | CA, BCL3 | Phase I | [123,124,125] |
| Checkpoint Inhibitors | Nivolumab | PD-1 | Phase II–III | [126,127,128,129,130,131] |
| Pembrolizumab | PD-1 | Phase I–II | [132,133,134] | |
| Ipilimumab | CTLA-4 | Phase I | [129] | |
| Diabetic Agents | Metformin | AMPK, Cl-Channels, mTOR | Phase I/II | [46,135,136,137,138,139,140,141,142,143,144,145,146,147] |
| Small Molecules and Biologics | Cetuximab | EGFR | Phase I | [148] |
| 5-ALA | Not Applicable | Phase III | [149,150,151,152] | |
| Cocktails | CUSP-9 | Survival Pathways | Phase I | [5,153,154,155,156,157] |
| CLOVA | GSK-3B | Phase I | [153] | |
| Celecoxib, Vinblastine, Cyclophosphamide | COX-2, Microtubules, DNA | Phase I | [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyne, S.B.; Yamini, B. An Alternative Pipeline for Glioblastoma Therapeutics: A Systematic Review of Drug Repurposing in Glioblastoma. Cancers 2021, 13, 1953. https://doi.org/10.3390/cancers13081953
Lyne SB, Yamini B. An Alternative Pipeline for Glioblastoma Therapeutics: A Systematic Review of Drug Repurposing in Glioblastoma. Cancers. 2021; 13(8):1953. https://doi.org/10.3390/cancers13081953
Chicago/Turabian StyleLyne, Seán B., and Bakhtiar Yamini. 2021. "An Alternative Pipeline for Glioblastoma Therapeutics: A Systematic Review of Drug Repurposing in Glioblastoma" Cancers 13, no. 8: 1953. https://doi.org/10.3390/cancers13081953
APA StyleLyne, S. B., & Yamini, B. (2021). An Alternative Pipeline for Glioblastoma Therapeutics: A Systematic Review of Drug Repurposing in Glioblastoma. Cancers, 13(8), 1953. https://doi.org/10.3390/cancers13081953