
A Novel Thienopyrimidine Analog, TPH104, Mediates Immunogenic Cell Death in Triple-Negative Breast Cancer Cells

 , , and
, , and 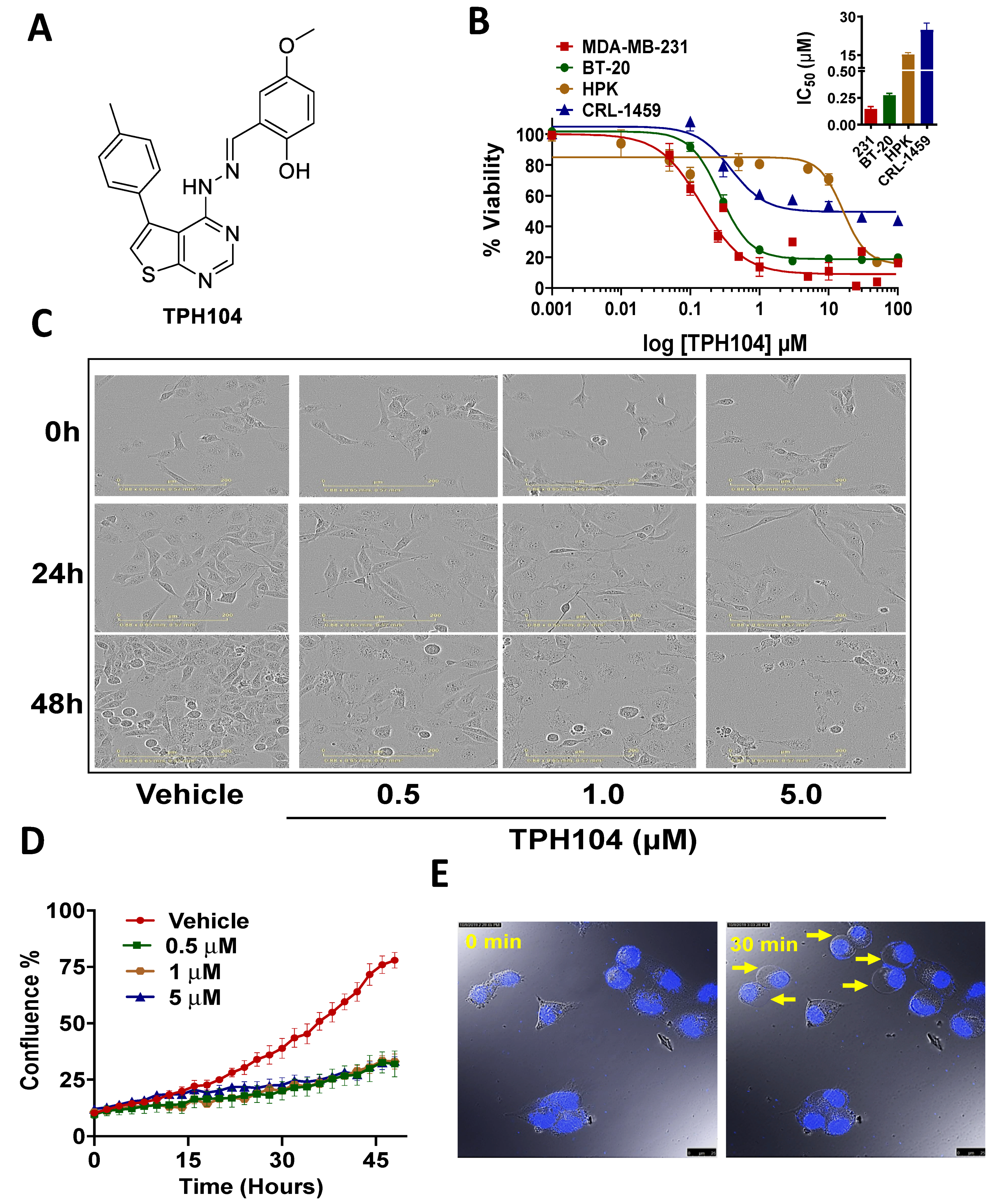{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Antibodies and Reagents
2.2. Cell Culture
2.3. Cell Cytotoxicity Assay
2.4. IncuCyte™ Live-Cell Morphology Study
2.5. Preparation of the Conditioned Media (CM) from Death-Primed MDA-MB-231 Cells
2.6. The Generation of Mouse Bone Marrow-Derived Dendritic Cells (BMDCs)
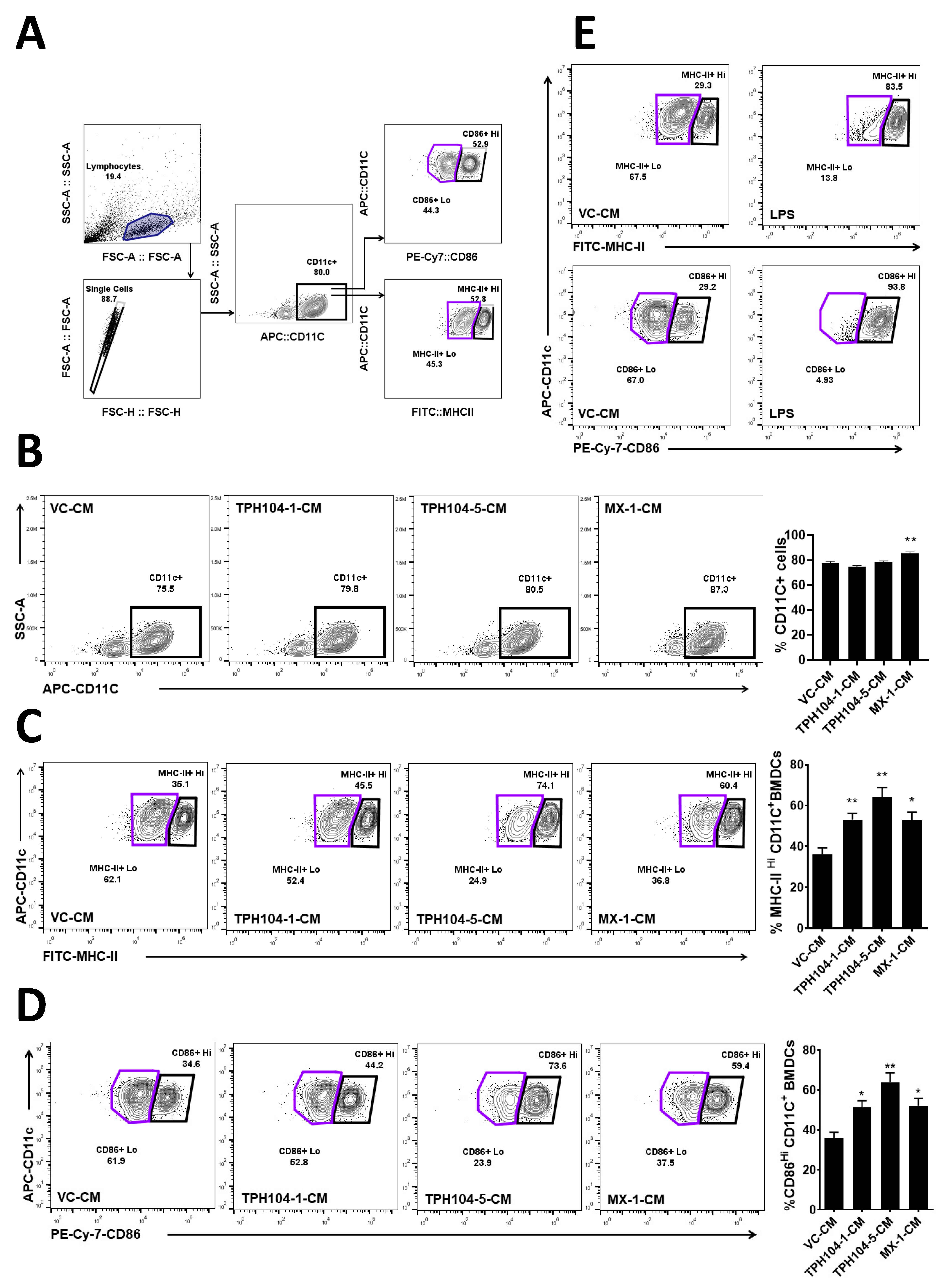
2.7. Flow Cytometry
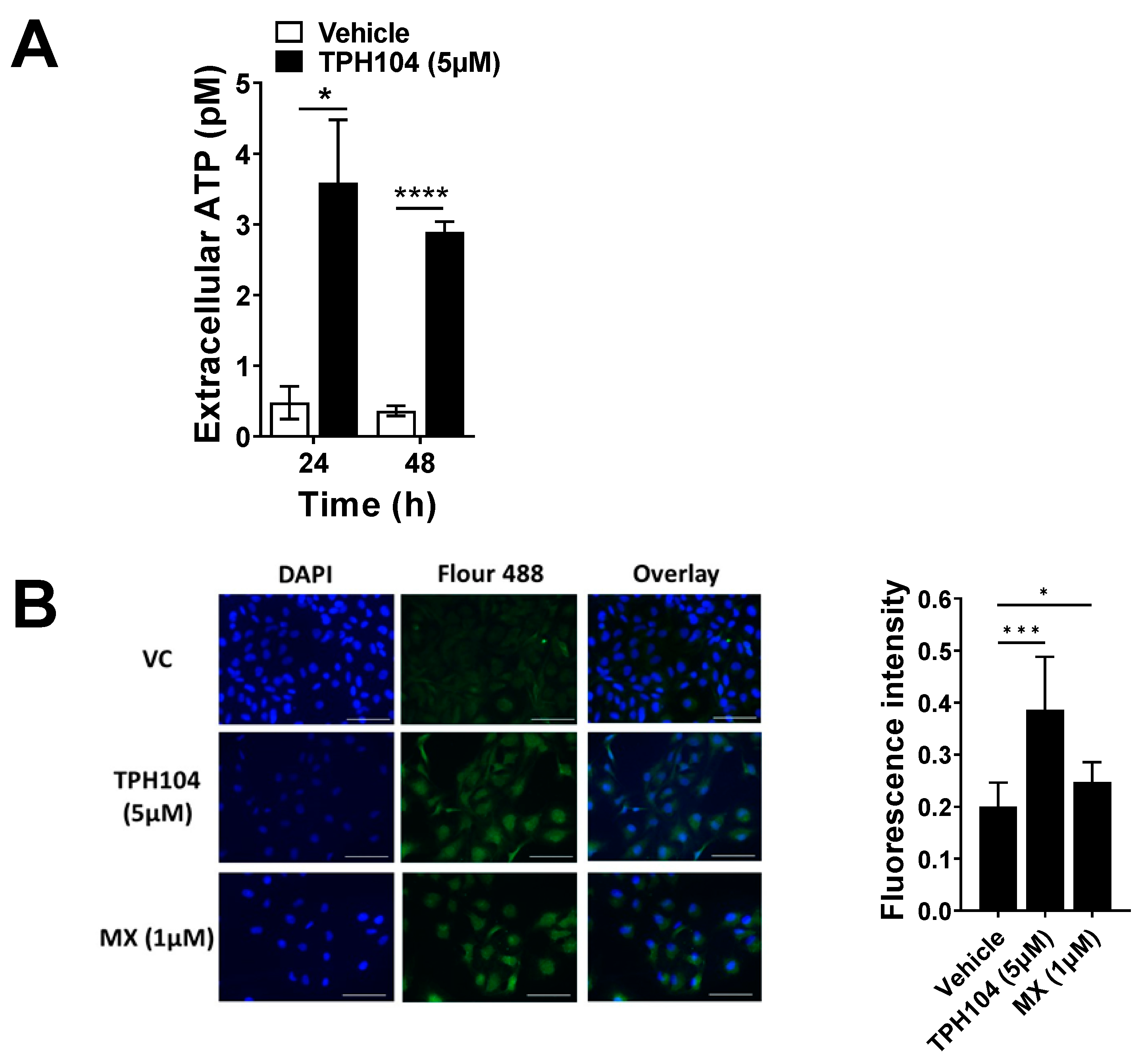
2.8. Extracellular ATP Release Measurements
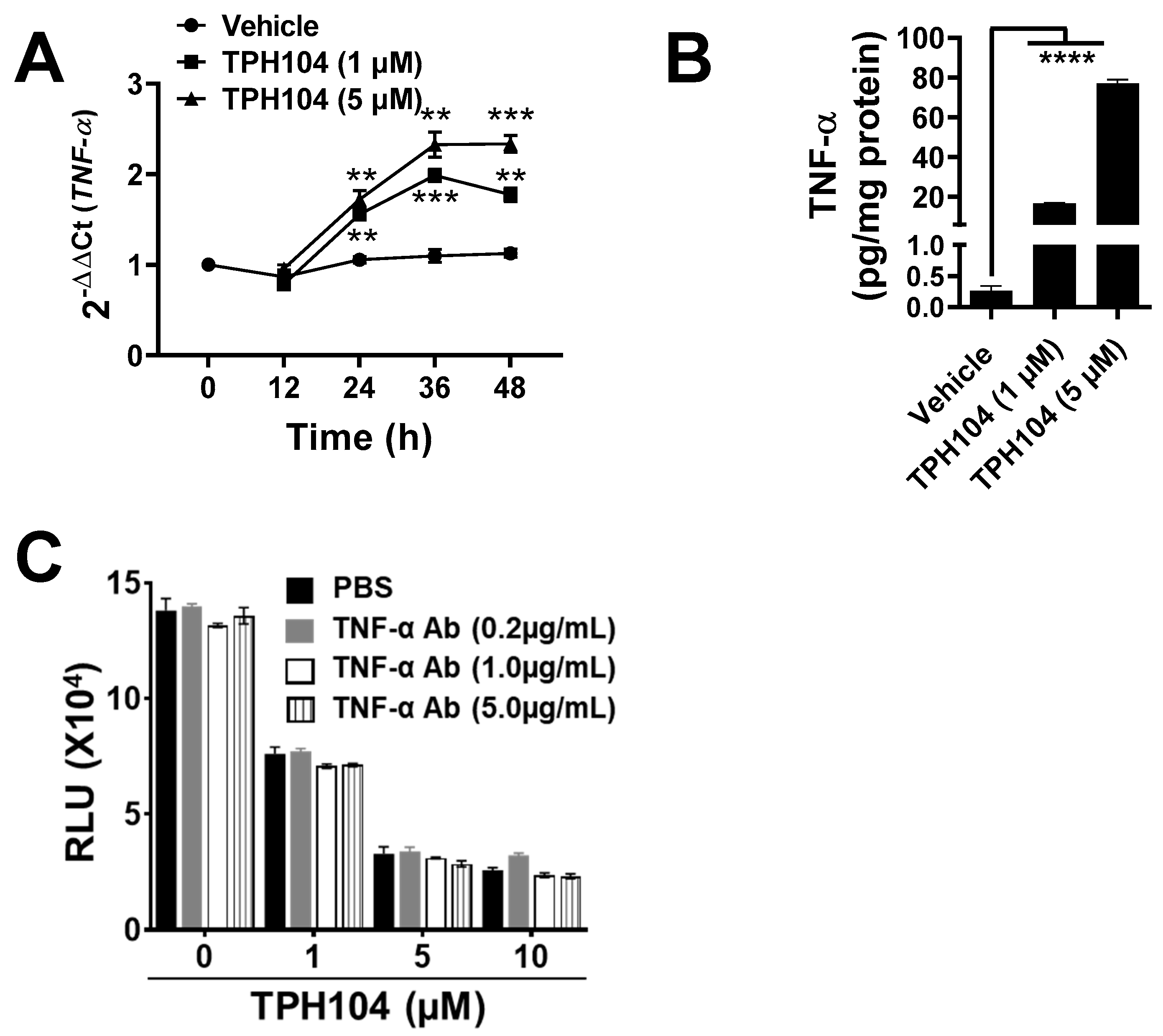
2.9. Measurement of TNF-α
2.10. RNA Extraction and Real-Time Quantitative PCR
2.11. Immunofluorescence for Calreticulin
2.12. Statistical Analysis
3. Results
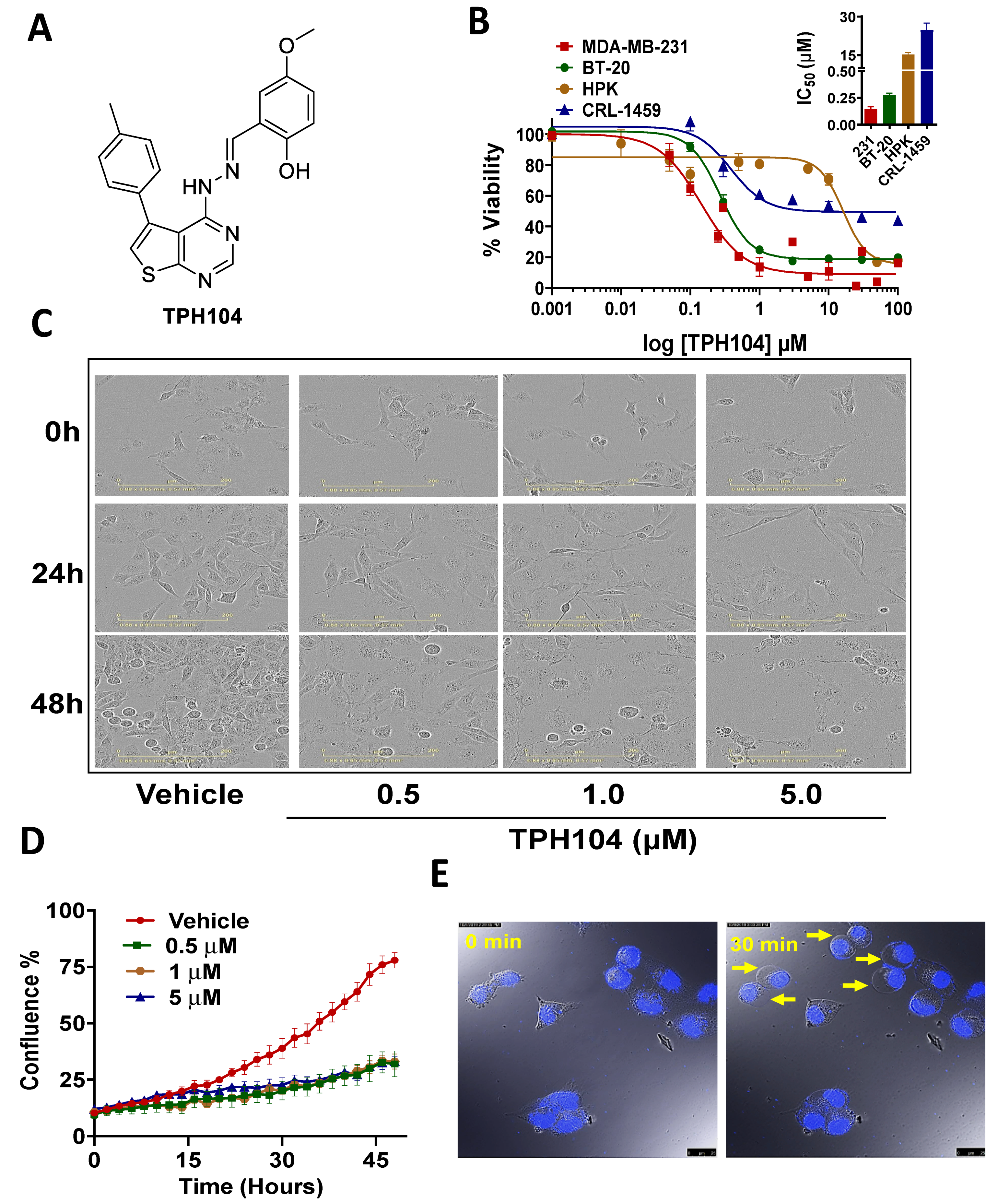
3.1. Cytotoxicity of TPH104 Is Selective to MDA-MB-231 Cells
3.2. TPH104-Mediated Death Primes MDA-MB-231 Cells to Express Biomarkers of Immunogenicity
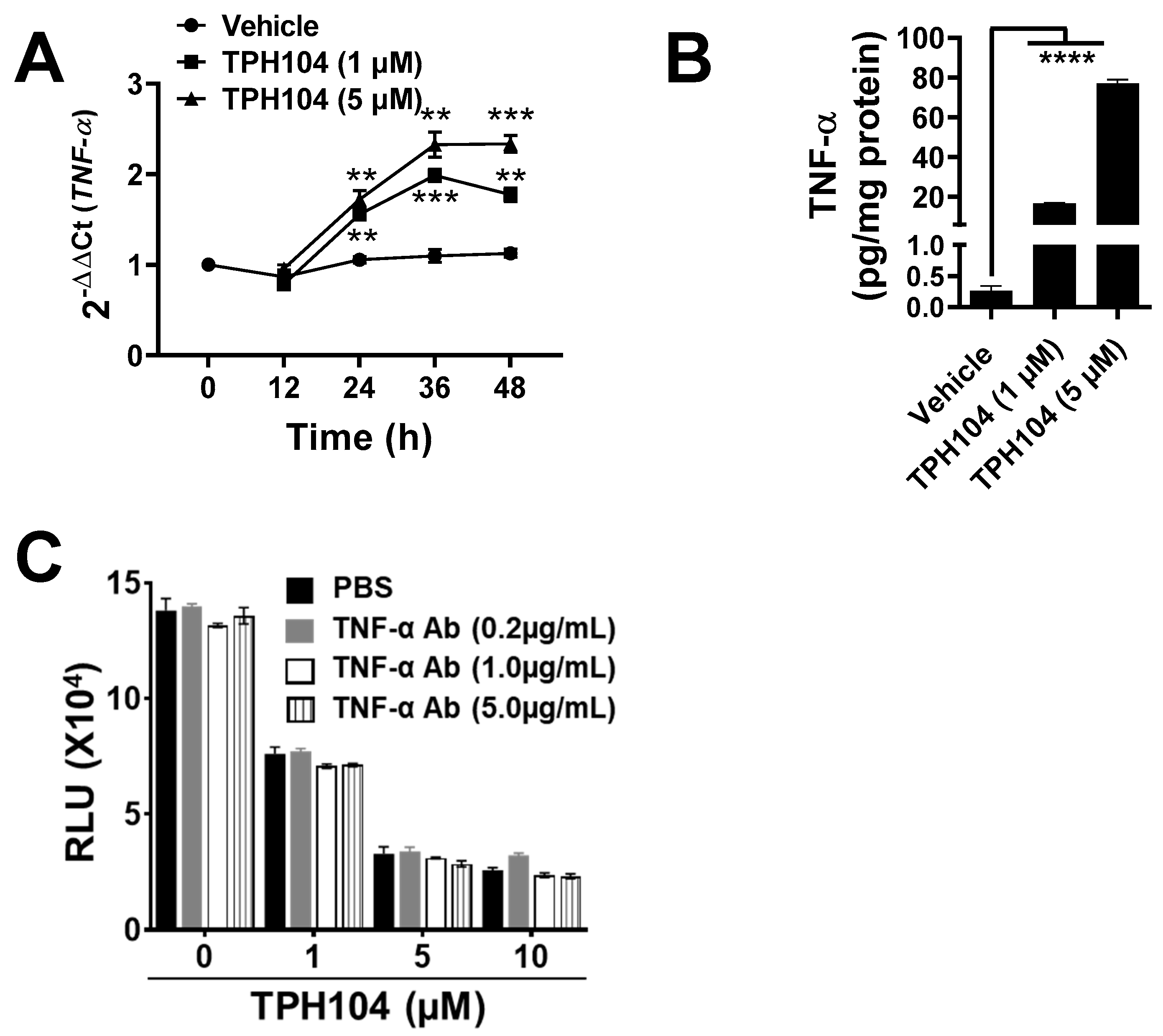
3.3. TPHI04 Induces the Production of TNF-α by MDA-MB-231 Cells
3.4. TPH104 Elicits the Release of Factors from MDA-MB-231 Cells That Induce the Maturation of DCs
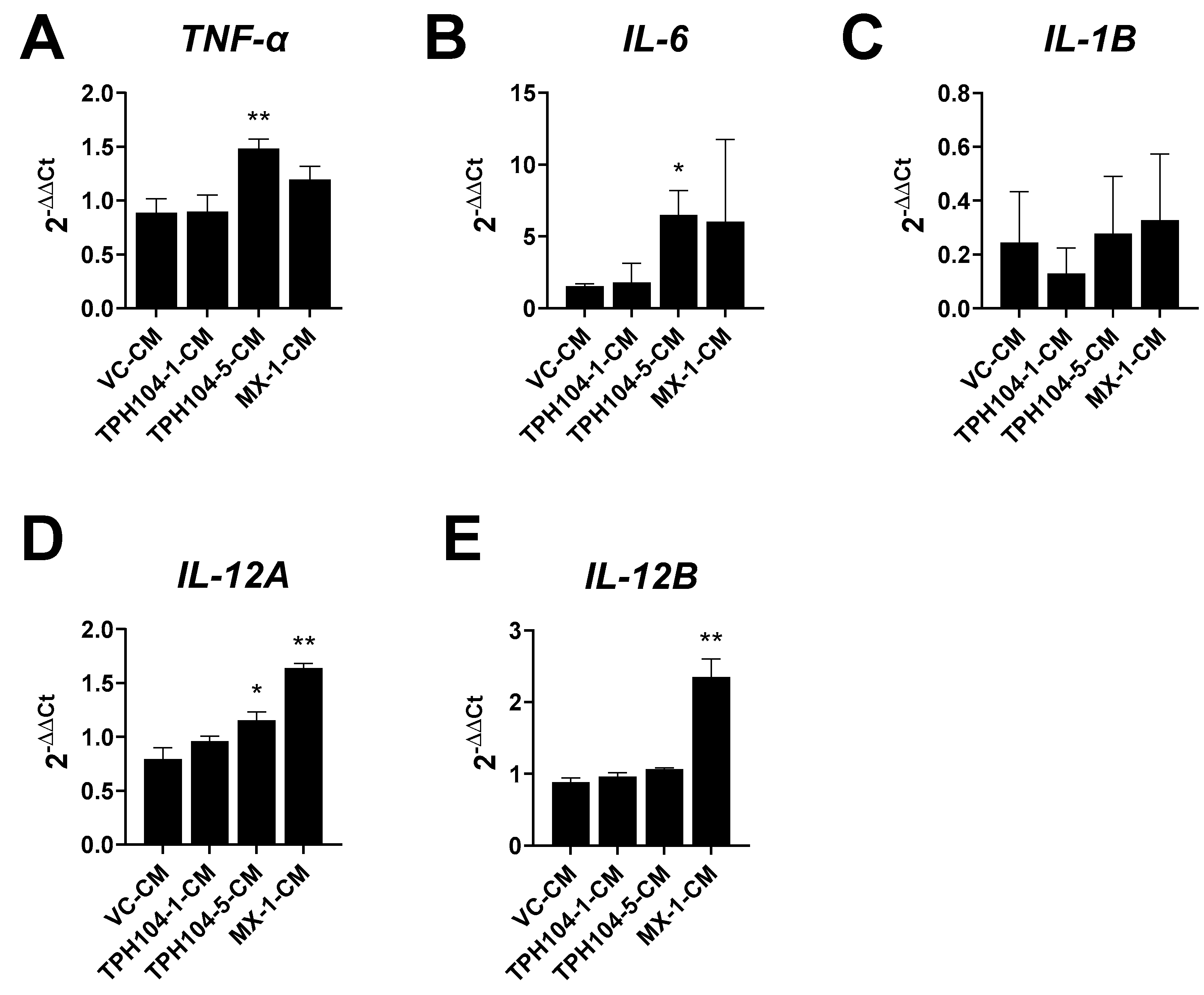
3.5. MDA-MB-231 Cells Stressed by TPH104 Release Factors to Modulate BMDC Cytokine Expression
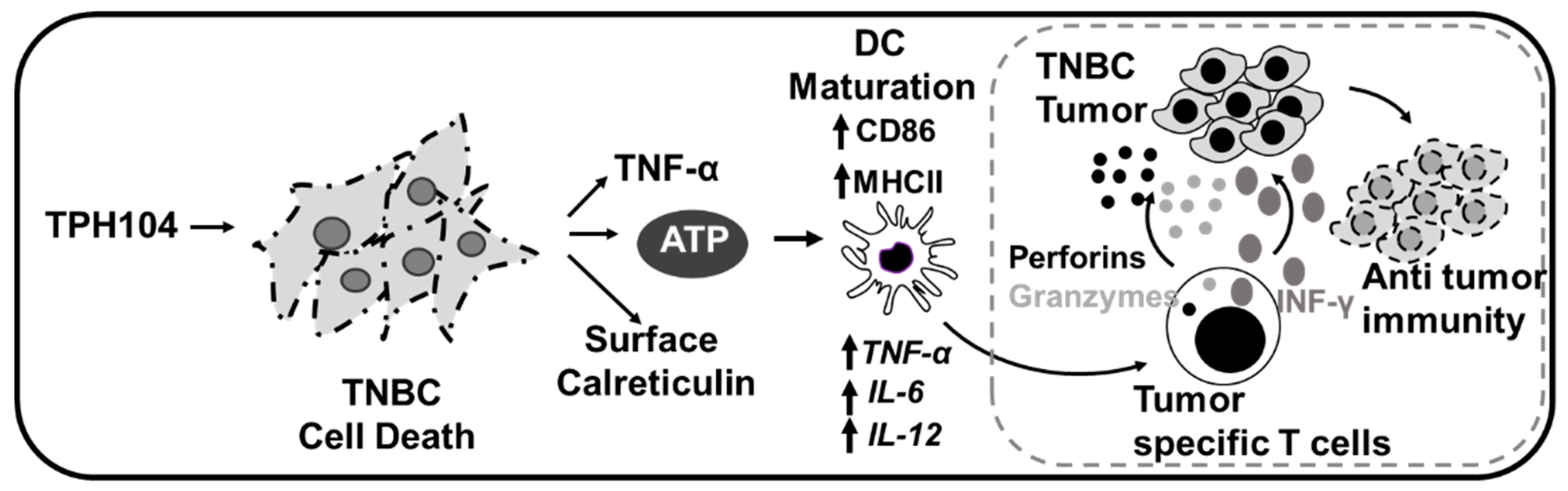
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Apetoh, L.; Tesniere, A.; Ghiringhelli, F.; Kroemer, G.; Zitvogel, L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. 2008, 68, 4026–4030. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spisek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Bajor, A.; Tischer, S.; Figueiredo, C.; Wittmann, M.; Immenschuh, S.; Blasczyk, R.; Eiz-Vesper, B. Modulatory role of calreticulin as chaperokine for dendritic cell-based immunotherapy. Clin. Exp. Immunol. 2011, 165, 220–234. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Michalak, M.; Opas, M.; Eggleton, P. The ins and outs of calreticulin: From the ER lumen to the extracellular space. Trends Cell Biol. 2001, 11, 122–129. [Google Scholar] [CrossRef]
- Inaba, K.; Turley, S.; Yamaide, F.; Iyoda, T.; Mahnke, K.; Inaba, M.; Pack, M.; Subklewe, M.; Sauter, B.; Sheff, D.; et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 1998, 188, 2163–2173. [Google Scholar] [CrossRef]
- Tufi, R.; Panaretakis, T.; Bianchi, K.; Criollo, A.; Fazi, B.; Di Sano, F.; Tesniere, A.; Kepp, O.; Paterlini-Brechot, P.; Zitvogel, L.; et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 2008, 15, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Bauer, D.; Mazzio, E.; Soliman, K.F. Whole transcriptomic analysis of apigenin on TNFα immuno-activated MDA-MB-231 breast cancer cells. Cancer Genom. Proteom. 2019, 16, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.; Nguyen, N. Mechanisms of tumor-induced T cell immune suppression and therapeutics to counter those effects. Arch. Pharm. Res. 2015, 38, 1415–1433. [Google Scholar] [CrossRef]
- Töpfer, K.; Kempe, S.; Müller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Qiu, Y.; Lu, W.; Jiang, Y.; Wang, J. Immunotherapeutic interventions of Triple Negative Breast Cancer. J. Transl. Med. 2018, 16, 147. [Google Scholar] [CrossRef] [Green Version]
- Wein, L.; Savas, P.; Luen, S.J.; Virassamy, B.; Salgado, R.; Loi, S. Clinical Validity and Utility of Tumor-Infiltrating Lymphocytes in Routine Clinical Practice for Breast Cancer Patients: Current and Future Directions. Front. Oncol. 2017, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Park, I.A.; Song, I.H.; Shin, S.J.; Kim, J.Y.; Yu, J.H.; Gong, G. Tertiary lymphoid structures: Prognostic significance and relationship with tumour-infiltrating lymphocytes in triple-negative breast cancer. J. Clin. Pathol. 2016, 69, 422–430. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.J.; Song, I.H.; Bang, W.S.; Heo, S.H.; Gong, G.; Park, I.A. CD11c-Positive Dendritic Cells in Triple-negative Breast Cancer. In Vivo 2018, 32, 1561–1569. [Google Scholar] [CrossRef] [Green Version]
- Gosavi, R.A.; Salwe, S.; Mukherjee, S.; Dahake, R.; Kothari, S.; Patel, V.; Chowdhary, A.; Deshmukh, R.A. Optimization of Ex Vivo Murine Bone Marrow Derived Immature Dendritic Cells: A Comparative Analysis of Flask Culture Method and Mouse CD11c Positive Selection Kit Method. Bone Marrow Res. 2018, 2018, 3495086. [Google Scholar] [CrossRef] [Green Version]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 2018, 16, 242. [Google Scholar] [CrossRef] [PubMed]
- de Almagro, M.C.; Goncharov, T.; Newton, K.; Vucic, D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 2015, 6, e1800. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Di Leo, A.; de Azambuja, E.; Larsimont, D.; Haibe-Kains, B.; Selleslags, J.; Delaloge, S.; Duhem, C.; Kains, J.P.; Carly, B.; et al. Multifactorial approach to predicting resistance to anthracyclines. J. Clin. Oncol. 2011, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Miragaya, J.; Palafox, M.; Pare, L.; Yoldi, G.; Ferrer, I.; Vila, S.; Galvan, P.; Pellegrini, P.; Perez-Montoyo, H.; Igea, A.; et al. Resistance to Taxanes in Triple-Negative Breast Cancer Associates with the Dynamics of a CD49f+ Tumor-Initiating Population. Stem Cell Rep. 2017, 8, 1392–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofferson, D.E.; Li, Y.; Hitomi, J.; Zhou, W.; Upperman, C.; Zhu, H.; Gerber, S.A.; Gygi, S.; Yuan, J. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012, 3, e320. [Google Scholar] [CrossRef]
- Dubar, G.; Azria, E.; Tesniere, A.; Dupont, H.; Le Ray, C.; Baugnon, T.; Matheron, S.; Luton, D.; Richard, J.C.; Launay, O.; et al. French experience of 2009 A/H1N1v influenza in pregnant women. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yang, K.; Zhao, R.; Ji, T.; Wang, X.; Yang, X.; Zhang, Y.; Cheng, K.; Liu, S.; Hao, J.; et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials 2016, 102, 187–197. [Google Scholar] [CrossRef]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol. Lett. 2016, 11, 2169–2175. [Google Scholar] [CrossRef] [Green Version]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4, 143ra199. [Google Scholar] [CrossRef] [Green Version]
- Dudek-Peric, A.M.; Ferreira, G.B.; Muchowicz, A.; Wouters, J.; Prada, N.; Martin, S.; Kiviluoto, S.; Winiarska, M.; Boon, L.; Mathieu, C.; et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res. 2015, 75, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Song, K.H.; Jung, S.Y.; Kang, S.M.; Kim, M.H.; Ahn, J.; Hwang, S.G.; Lee, J.H.; Lim, D.S.; Nam, S.Y.; Song, J.Y. Induction of immunogenic cell death by radiation-upregulated karyopherin alpha 2 in vitro. Eur. J. Cell Biol. 2016, 95, 219–227. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Karthikeyan, C.; Nyinawabera, A. Necroptosis Inducers or Autophagy Inhibitors or a Combination Hereof. World Intellectual Property Organization. WO2019200221A1, 2019. PCT/US2019/027167. [Google Scholar]
- Amawi, H.; Hussein, N.; Boddu, S.H.S.; Karthikeyan, C.; Williams, F.E.; Ashby, C.R., Jr.; Raman, D.; Trivedi, P.; Tiwari, A.K. Novel Thienopyrimidine Derivative, RP-010, Induces beta-Catenin Fragmentation and Is Efficacious against Prostate Cancer Cells. Cancers 2019, 11, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Oudat, B.A.; Ramapuram, H.; Malla, S.; Audat, S.A.; Hussein, N.; Len, J.M.; Kumari, S.; Bedi, M.F.; Ashby, C.R.; Tiwari, A.K. Novel Chrysin-De-Allyl PAC-1 Hybrid Analogues as Anticancer Compounds: Design, Synthesis, and Biological Evaluation. Molecules 2020, 25, 3063. [Google Scholar] [CrossRef] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]
- Diwakar, B.T.; Yoast, R.; Nettleford, S.; Qian, F.; Lee, T.J.; Berry, S.; Huffnagle, I.; Rossi, R.M.; Trebak, M.; Paulson, R.F.; et al. Crth2 receptor signaling down-regulates lipopolysaccharide-induced NF-kappaB activation in murine macrophages via changes in intracellular calcium. FASEB J. 2019, fj201802608R. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Trombetta, E.S.; Ebersold, M.; Garrett, W.; Pypaert, M.; Mellman, I. Activation of lysosomal function during dendritic cell maturation. Science 2003, 299, 1400–1403. [Google Scholar] [CrossRef]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 2016, 30, 651. [Google Scholar] [CrossRef]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef]
- Groenendyk, J.; Lynch, J.; Michalak, M. Calreticulin, Ca2+, and calcineurin - signaling from the endoplasmic reticulum. Mol. Cells 2004, 17, 383–389. [Google Scholar]
- Holaska, J.M.; Black, B.E.; Rastinejad, F.; Paschal, B.M. Ca2+-dependent nuclear export mediated by calreticulin. Mol. Cell Biol. 2002, 22, 6286–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldolesi, J.; Krause, K.H.; Michalak, M. Calreticulin: How many functions in how many cellular compartments? Como, April 1996. Cell Calcium 1996, 20, 83–86. [Google Scholar] [CrossRef]
- Coppolino, M.G.; Woodside, M.J.; Demaurex, N.; Grinstein, S.; St-Arnaud, R.; Dedhar, S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 1997, 386, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Cariello, N.F.; Keohavong, P.; Kat, A.G.; Thilly, W.G. Molecular analysis of complex human cell populations: Mutational spectra of MNNG and ICR-191. Mutat. Res. 1990, 231, 165–176. [Google Scholar] [CrossRef]
- Li, Z.L.; Zhang, H.L.; Huang, Y.; Huang, J.H.; Sun, P.; Zhou, N.N.; Chen, Y.H.; Mai, J.; Wang, Y.; Yu, Y.; et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat. Commun. 2020, 11, 3806. [Google Scholar] [CrossRef]
- Boyd-Tressler, A.; Penuela, S.; Laird, D.W.; Dubyak, G.R. Chemotherapeutic drugs induce ATP release via caspase-gated pannexin-1 channels and a caspase/pannexin-1-independent mechanism. J. Biol. Chem. 2014, 289, 27246–27263. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, A.P. Anoikis. Cell Death Differ 2005, 12 Suppl. 2, 1473–1477. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rivandi, M.; Abedini, S.; Pasdar, A.; Sahebkar, A. Regulators and mechanisms of anoikis in triple-negative breast cancer (TNBC): A review. Crit. Rev. Oncol. Hematol. 2019, 140, 17–27. [Google Scholar] [CrossRef]
- Orozco, S.L.; Daniels, B.P.; Yatim, N.; Messmer, M.N.; Quarato, G.; Chen-Harris, H.; Cullen, S.P.; Snyder, A.G.; Ralli-Jain, P.; Frase, S. RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Rep. 2019, 28, 2275–2287.e5. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, S.; Herfs, M.; Delvenne, P.; Hubert, P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: Insight into the molecular mechanisms. J. Leukoc. Biol. 2013, 93, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8⁺ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef] [PubMed]
- Brunner, C.; Seiderer, J.; Schlamp, A.; Bidlingmaier, M.; Eigler, A.; Haimerl, W.; Lehr, H.A.; Krieg, A.M.; Hartmann, G.; Endres, S. Enhanced dendritic cell maturation by TNF-alpha or cytidine-phosphate-guanosine DNA drives T cell activation in vitro and therapeutic anti-tumor immune responses in vivo. J. Immunol. 2000, 165, 6278–6286. [Google Scholar] [CrossRef] [Green Version]
- den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 2008, 181, 5396–5404. [Google Scholar] [CrossRef]
- den Haan, J.M.; Bevan, M.J. A novel helper role for CD4 T cells. Proc. Natl. Acad. Sci. USA 2000, 97, 12950–12952. [Google Scholar] [CrossRef] [Green Version]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. The interleukin-12/interleukin-12-receptor system: Role in normal and pathologic immune responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef]
- Murphy, K.M.; Reiner, S.L. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2002, 2, 933–944. [Google Scholar] [CrossRef]
- Martin-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Sgadari, C.; Angiolillo, A.L.; Tosato, G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood 1996, 87, 3877–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Cheng, P.; Mottram, P.; Alvarez, X.; Moons, L.; Evdemon-Hogan, M.; Wei, S.; Zou, L.; Kryczek, I.; Hoyle, G.; et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004, 64, 5535–5538. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tukaramrao, D.B.; Malla, S.; Saraiya, S.; Hanely, R.A.; Ray, A.; Kumari, S.; Raman, D.; Tiwari, A.K. A Novel Thienopyrimidine Analog, TPH104, Mediates Immunogenic Cell Death in Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 1954. https://doi.org/10.3390/cancers13081954
Tukaramrao DB, Malla S, Saraiya S, Hanely RA, Ray A, Kumari S, Raman D, Tiwari AK. A Novel Thienopyrimidine Analog, TPH104, Mediates Immunogenic Cell Death in Triple-Negative Breast Cancer Cells. Cancers. 2021; 13(8):1954. https://doi.org/10.3390/cancers13081954
Chicago/Turabian StyleTukaramrao, Diwakar Bastihalli, Saloni Malla, Siddharth Saraiya, Ross Allen Hanely, Aniruddha Ray, Shikha Kumari, Dayanidhi Raman, and Amit K. Tiwari. 2021. "A Novel Thienopyrimidine Analog, TPH104, Mediates Immunogenic Cell Death in Triple-Negative Breast Cancer Cells" Cancers 13, no. 8: 1954. https://doi.org/10.3390/cancers13081954