
CAR-T in Cancer Treatment: Develop in Self-Optimization, Win-Win in Cooperation


Abstract
:Simple Summary
Abstract
1. Introduction
2. Enhanced Persistence and Antitumor Properties of CAR-T Therapy
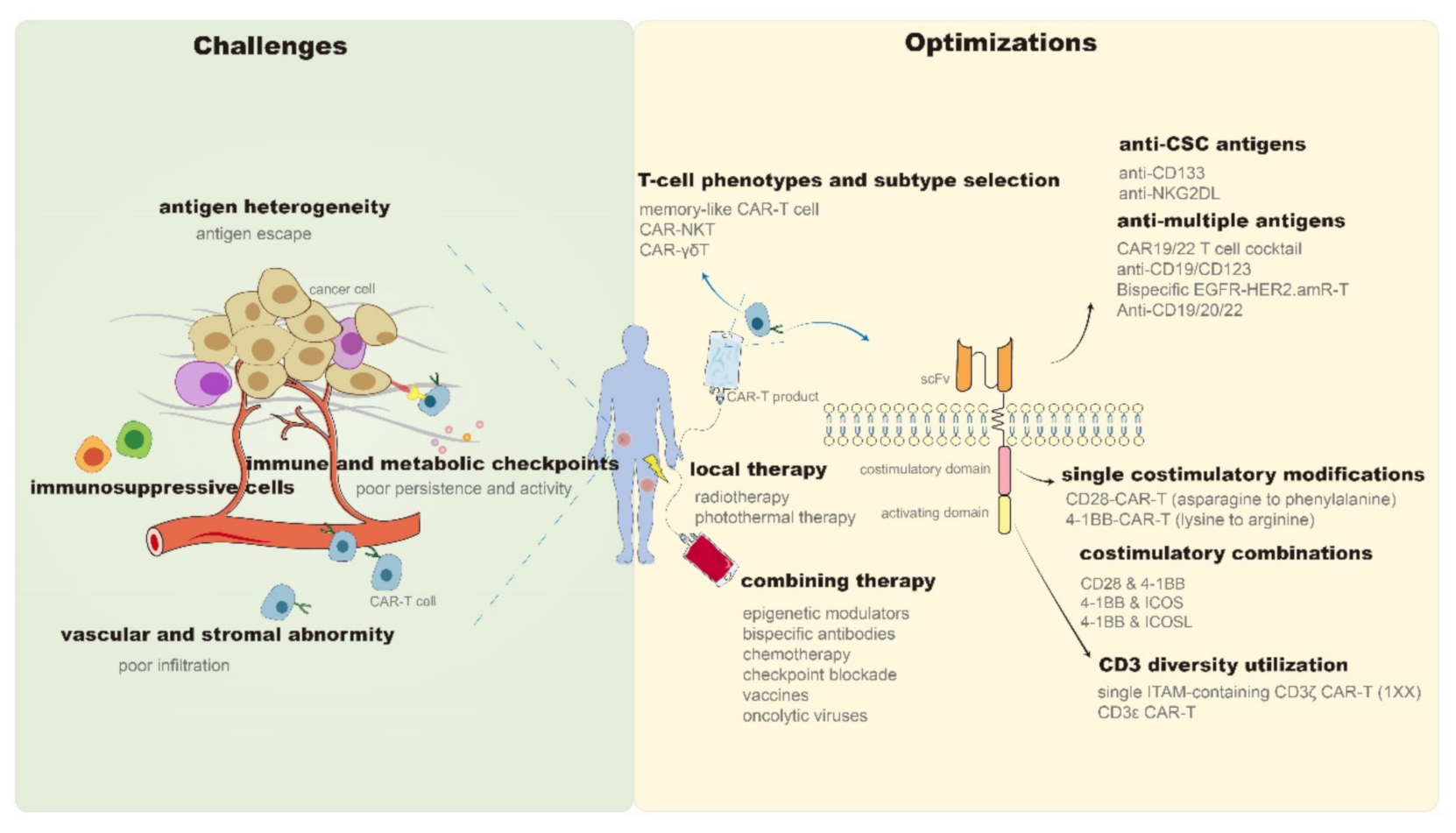
2.1. Optimization of CAR-T Structures
2.1.1. Optimization of the Extracellular Domain
Optimization of the Extracellular Domain
Targeting Simultaneously on Multiple Antigens
2.1.2. Optimization of the Intracellular Domain
Improving Intracellular Activating Domain
Improving Intracellular Co-Stimulatory Domain
2.1.3. Optimization of T Cell Selection
2.2. Combined Treatment Strategies of CAR-T
2.2.1. Combination Strategies to Enhance Antigen Recognition
Combining CAR-T with Epigenetic Modulators
Combining CAR-T with Bispecific Antibodies
2.2.2. Combination Strategies to Overcome the Suppressive TME
Combining CAR-T with Chemotherapy
Combining CAR-T with Local Therapy
Combining CAR-T with Checkpoint Blockade
2.2.3. Combining CAR-T with Vaccines to Promote In Vivo Expansion
2.2.4. Combining CAR-T with Oncolytic Viruses to Deal with Solid-Tumor Challenges
3. Enhanced Clinical Safety and Accessibility of CAR-T Therapy
3.1. Safety Maintenance of CAR-T Therapy
3.1.1. The Endogenous Witches
3.1.2. The Exogenous Switches
3.2. Accessibility Increase of CAR-T Therapy
3.2.1. Acquisition of T Cells
3.2.2. Optimization of CAR-T Manufacturing Process
4. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheridan, C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat. Biotechnol. 2017, 35, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Go, W.Y.; Neelapu, S.S. Development and Use of the Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy Axicabtagene Ciloleucel in Large B-Cell Lymphoma A Review. JAMA Oncol. 2020, 6, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T Cells into Tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Espat, N.J.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budhu, S.; Schaer, D.A.; Li, Y.; Toledo-Crow, R.; Panageas, K.; Yang, X.; Zhong, H.; Houghton, A.N.; Silverstein, S.C.; Merghoub, T.; et al. Blockade of surface-bound TGF-beta on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal. 2017, 10, eaak9702. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells in vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef]
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of Tumor Destruction by Chimeric Antigen Receptor-modified T Cells. Mol. Ther. 2014, 22, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Lesch, S.; Benmebarek, M.-R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Biol. 2020, 65, 80–90. [Google Scholar] [CrossRef]
- Chou, C.K.; Turtle, C.J. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant. 2019, 54, 780–784. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Sadeghi, S.; Fakhr, E.; Abazari, M.F.; Poortahmasebi, V.; Kheirollahi, A.; Askari, H.; Rajabzadeh, A.; Rastegarpanah, M.; Linē, A.; et al. Production of CAR T-cells by GMP-grade lentiviral vectors: Latest advances and future prospects. Crit. Rev. Clin. Lab. Sci. 2019, 56, 393–419. [Google Scholar] [CrossRef]
- Gee, A.P. GMP CAR-T cell production. Best Pract. Res. Clin. Haematol. 2018, 31, 126–134. [Google Scholar] [CrossRef]
- Dai, X.; Mei, Y.; Cai, D.; Han, W. Standardizing CAR-T therapy: Getting it scaled up. Biotechnol. Adv. 2019, 37, 239–245. [Google Scholar] [CrossRef]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell 2012, 21, 283–296.e4. [Google Scholar] [CrossRef] [Green Version]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef]
- Wang, L.; Yin, E.T.S.; Zhao, H.; Ni, F.; Hu, Y.; Huang, H. CAR-T cells: The Chinese experience. Expert Opin. Biol. Ther. 2020, 20, 1293–1308. [Google Scholar] [CrossRef]
- Wang, N.; Hu, X.L.; Cao, W.Y.; Li, C.R.; Xiao, Y.; Cao, Y.; Gu, C.J.; Zhang, S.K.; Chen, L.T.; Cheng, J.L.; et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood 2020, 135, 17–27. [Google Scholar] [CrossRef]
- Pan, J.; Zuo, S.; Deng, B.; Xu, X.; Li, C.; Zheng, Q.; Ling, Z.; Song, W.; Xu, J.; Duan, J.; et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood 2020, 135, 387–391. [Google Scholar] [CrossRef]
- Dannenfelser, R.; Allen, G.M.; VanderSluis, B.; Koegel, A.K.; Levinson, S.; Stark, S.R.; Yao, V.; Tadych, A.; Troyanskaya, O.G.; Lim, W.A. Discriminatory Power of Combinatorial Antigen Recognition in Cancer T Cell Therapies. Cell Syst. 2020, 11, 215–228.e5. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Li, J.; Sun, C.; Gao, K.; Hirabayashi, K.; Li, H.; Savoldo, B.; Liu, R.; Dotti, G. Cancer Immunotherapy with T Cells Carrying Bispecific Receptors That Mimic Antibodies. Cancer Immunol. Res. 2019, 7, 773–783. [Google Scholar] [CrossRef]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia 2021, 35, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Wen, Q.; Zhong, J.F.; Zhang, C.; Zhang, X.; et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor–CD3 complex. Nat. Cell Biol. 2019, 573, 546–552. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef] [PubMed]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhou, Q.; Masubuchi, T.; Shi, X.; Li, H.; Xu, X.; Huang, M.; Meng, L.; He, X.; Zhu, H.; et al. Multiple Signaling Roles of CD3epsilon and Its Application in CAR-T Cell Therapy. Cell 2020, 182, 855–871.e23. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.M.; Scherer, L.D.; Rouce, R.H. Replacing CAR-T cell resistance with persistence by changing a single residue. J. Clin. Investig. 2020, 130, 2806–2808. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e6. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e6. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; Mcgettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Huang, X.; Huang, X.; Chen, W.; Hao, L.; Chen, Z. Chimeric antigen receptor modified T cell (CAR-T) co-expressed with ICOSL-41BB promote CAR-T proliferation and tumor rejection. Biomed. Pharmacother. 2019, 118, 109333. [Google Scholar] [CrossRef]
- Sheih, A.; Voillet, V.; Hanafi, L.-A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Kondo, T.; Ando, M.; Nagai, N.; Tomisato, W.; Srirat, T.; Liu, B.; Mise-Omata, S.; Ikeda, M.; Chikuma, S.; Nishimasu, H.; et al. The NOTCH–FOXM1 Axis Plays a Key Role in Mitochondrial Biogenesis in the Induction of Human Stem Cell Memory–like CAR-T Cells. Cancer Res. 2019, 80, 471–483. [Google Scholar] [CrossRef]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Anderson, J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Abramowski, P.; Don, N.D.W.; Flutter, B.; Capsomidis, A.; Cheung, G.W.-K.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef] [Green Version]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef]
- Driouk, L.; Gicobi, J.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Blood 2019, 134. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. OncoImmunology 2018, 7, e1368604. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2018, 79, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Sun, C.; Bernatchez, C.; Xia, X.; Hwu, P.; Dotti, G.; Li, S. T-cell Homing Therapy for Reducing Regulatory T Cells and Preserving Effector T-cell Function in Large Solid Tumors. Clin. Cancer Res. 2018, 24, 2920–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Feng, K.-C.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.-M.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor–Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef] [Green Version]
- Murty, S.; Haile, S.T.; Beinat, C.; Aalipour, A.; Alam, I.S.; Murty, T.; Shaffer, T.M.; Patel, C.B.; Graves, E.E.; Mackall, C.L.; et al. Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. OncoImmunology 2020, 9, 1757360. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, Q.Y.; Dukhovlinova, E.; Chen, G.J.; Ahn, S.; Wang, C.; Ogunnaike, E.A.; Ligler, F.S.; Dotti, G.; Gu, Z. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv. Mater. 2019, 31, 1900192. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Xu, X.; Gnanaprakasam, J.N.R.; Sherman, J.; Wang, R. A Metabolism Toolbox for CAR T Therapy. Front. Oncol. 2019, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Caruana, I.; Weber, G.; Ballard, B.C.; Wood, M.S.; Savoldo, B.; Dotti, G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo. Clin. Cancer Res. 2015, 21, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Zhang, L.; Zhang, H.; Ning, J.; Tu, S.; He, Y.; Li, Y. CD19 chimeric antigen receptor–redirected T cells combined with epidermal growth factor receptor pathway substrate 8 peptide–derived dendritic cell vaccine in leukemia. Cytotherapy 2019, 21, 659–670. [Google Scholar] [CrossRef]
- Ma, L.; Dichwalkar, T.; Chang, J.Y.H.; Cossette, B.; Garafola, D.; Zhang, A.Q.; Fichter, M.; Wang, C.; Liang, S.; Silva, M.; et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019, 365, 162–168. [Google Scholar]
- Park, A.K.; Fong, Y.; Kim, S.-I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.-C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, K.; Shaw, A.R.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus–Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef] [Green Version]
- Goebeler, M.-E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, E.; Jafarzadeh, L.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; Hadjati, J. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J. Exp. Clin. Cancer Res. 2020, 39, 1–12. [Google Scholar] [CrossRef]
- Yazdanifar, M.; Zhou, R.; Grover, P.; Williams, C.; Bose, M.; Moore, L.J.; Wu, S.T.; Maher, J.; Dreau, D.; Mukherjee, P. Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1070. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; Van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nat. Cell Biol. 2014, 507, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy (vol 18, pg 498, 2018). Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar]
- Guedan, S.; Alemany, R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef] [Green Version]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Penack, O.; Koenecke, C. Complications after CD19+ CAR T-Cell Therapy. Cancers 2020, 12, 3445. [Google Scholar] [CrossRef]
- Oved, J.H.; Barrett, D.M.; Teachey, D.T. Cellular therapy: Immune-related complications. Immunol. Rev. 2019, 290, 114–126. [Google Scholar] [CrossRef]
- Xu, X.-J.; Tang, Y.-M. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014, 343, 172–178. [Google Scholar] [CrossRef]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell–Associated Neurotoxicity. Front. Immunol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Schubert, M.-L.; Schmitt, M.; Wang, L.; Ramos, C.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 1–13. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Farooq, M.A.; Gao, Y.X.; Zhang, L.; Niu, C.Y.; Ajmal, I.; Zhou, Y.; He, C.; Zhao, G.X.; Yao, J.; et al. CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19(+)HLA-C1(-) Malignant B Cells While Sparing CD19(+)HLA-C1(+) Healthy B Cells. Cancers 2020, 12, 2612. [Google Scholar] [CrossRef] [PubMed]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e8. [Google Scholar] [CrossRef] [Green Version]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Puhringer, D.; Salzer, B.; Djinovic-Carugo, K.; Steinberger, P.; Linhares, A.D.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef]
- Giordano-Attianese, G.; Gainza, P.; Gray-Gaillard, E.; Cribioli, E.; Shui, S.; Kim, S.; Kwak, M.-J.; Vollers, S.; Osorio, A.D.J.C.; Reichenbach, P.; et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat. Biotechnol. 2020, 38, 426–432. [Google Scholar] [CrossRef]
- Sommer, C.; Cheng, H.-Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a "safety switch" to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Murty, S.; Labanieh, L.; Murty, T.; Gowrishankar, G.; Haywood, T.; Alam, I.S.; Beinat, C.; Robinson, E.; Aalipour, A.; Klysz, D.D.; et al. PET Reporter Gene Imaging and Ganciclovir-Mediated Ablation of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2020, 80, 4731–4740. [Google Scholar] [CrossRef]
- Richman, S.A.; Wang, L.-C.; Moon, E.K.; Khire, U.R.; Albelda, S.M.; Milone, M.C. Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo. Mol. Ther. 2020, 28, 1600–1613. [Google Scholar] [CrossRef]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Loureiro, L.R.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. OncoImmunology 2020, 9, 1785608. [Google Scholar] [CrossRef]
- Albert, S.; Arndt, C.; Feldmann, A.; Bergmann, R.; Bachmann, D.; Koristka, S.; Ludwig, F.; Ziller-Walter, P.; Kegler, A.; Gärtner, S.; et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. OncoImmunology 2017, 6, e1287246. [Google Scholar] [CrossRef]
- Kim, M.S.; Ma, J.S.Y.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef]
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.-H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef] [Green Version]
- Roddie, C.; O’Reilly, M.; Pinto, J.D.A.; Vispute, K.; Lowdell, M. Manufacturing chimeric antigen receptor T cells: Issues and challenges. Cytotherapy 2019, 21, 327–340. [Google Scholar] [CrossRef]
- Kacherovsky, N.; Cardle, I.I.; Cheng, E.L.; Yu, J.L.; Baldwin, M.L.; Salipante, S.J.; Jensen, M.C.; Pun, S.H. Traceless aptamer-mediated isolation of CD8+ T cells for chimeric antigen receptor T-cell therapy. Nat. Biomed. Eng. 2019, 3, 783–795. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- From Pluripotent Stem to CAR T Cells. Cancer Discov. 2018, 8, OF5. [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, K.M.C.M.; Sadelain, J.E.J.M.-S.T.G.S.J.C.V.D.S.M.H.A.O.M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nat. Cell Biol. 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Chang, C.; Van Der Stegen, S.; Mili, M.; Clarke, R.; Lai, Y.-S.; Witty, A.; Lindenbergh, P.; Yang, B.-H.; Husain, M.; Shaked, H.; et al. FT819: Translation of Off-the-Shelf TCR-Less Trac-1XX CAR-T Cells in Support of First-of-Kind Phase I Clinical Trial. Blood 2019, 134, 4434. [Google Scholar] [CrossRef]
- Ferreira, M.V.; Cabral, E.T.; Coroadinha, A.S. Progress and Perspectives in the Development of Lentiviral Vector Producer Cells. Biotechnol. J. 2021, 16. [Google Scholar] [CrossRef]
- Harris, E.; Elmer, J.J. Optimization of electroporation and other non-viral gene delivery strategies for T cells. Biotechnol. Prog. 2021, 37, 37. [Google Scholar] [CrossRef]
- Bishop, D.C.; Caproni, L.; Gowrishankar, K.; Legiewicz, M.; Karbowniczek, K.; Tite, J.; Gottlieb, D.J.; Micklethwaite, K.P. CAR T Cell Generation by piggyBac Transposition from Linear Doggybone DNA Vectors Requires Transposon DNA-Flanking Regions. Mol. Ther. Methods Clin. Dev. 2020, 17, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z.; et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019, 37, 1502–1512. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, Y.; Yang, J.; Li, W.; Tian, Y.; Wei, G.; Zhang, L.; Zhao, K.; Qi, Y.; Tan, B.; et al. Development and clinical evaluation of non-viral genome specific targeted CAR T cells in relapsed/refractory B-cell non-Hodgkin lymphoma. medRxiv 2020. [Google Scholar] [CrossRef]
- Dai, X.; Park, J.J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV–Cpf1. Nat. Methods 2019, 16, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Moffett, H.F.; Coon, M.E.; Radtke, S.; Stephan, S.B.; McKnight, L.; Lambert, A.; Stoddard, B.L.; Kiem, H.P.; Stephan, M.T. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Combined Strategy | Impact | Tumor Model | Refs. |
|---|---|---|---|
| Combined Strategies to Enhance Antigen Recognition | |||
| Combining CAR-T with Epigenetic Modulators | |||
| Decitabine & MUC1-CAR-T | Upregulate MUC1 expression | Pancreatic cancer | [12] |
| Valproic acid & NKG2D-CAR-T | Upregulate NKG2D-ligand expression | AML | [48] |
| GSIs & BCMA-CAR-T | Increase BCMA and decrease sBCMA | MM | [49] |
| Combining CAR-T with Bispecific Antibodies | |||
| EGFR-BiTE & EGFRvIII-CAR-T | Eradicate heterogeneous tumor cells | Glioblastoma | [50] |
| Biotinylated bispecific antibody & CAR-T | Mediate tumor lysis and IFN-γ production | Different tumors | [51] |
| Cocktail of fluorescein-bispecific adapters & CAR-T | Eradicate heterogeneous tumor cells | Solid tumors | [52] |
| Combined Strategies to Overcome the Suppressive TME | |||
| Combining CAR-T with Chemotherapy | |||
| IL-12 plus doxorubicin & CAR-T | Increase T-cell deep penetration | Solid tumors | [53] |
| Nab-paclitaxel plus cyclophosphamide & CAR-T | Promote CAR-T infiltration | Biliary tract cancer | [54] |
| Combining CAR-T with local therapy | |||
| Focal radiotherapy & GD2 CAR-T | Promote CAR-T extravasation and expansion | Glioblastoma | [55] |
| Photothermal therapy & CAR-T | Promote CAR-T accumulation | Solid tumors | [56] |
| Combining CAR-T with checkpoint blockade | |||
| Immune checkpoint blockade & CAR-T | Maximize CAR-T efficacy | Solid tumors | [57] |
| Metabolic checkpoint blockade & CAR-T | Maximize CAR-T efficacy | Solid tumors | [58] |
| Combined strategies to promote in vivo expansion | |||
| Whole-cell vaccine (tumor/DC) & CAR-T | Boost CAR-T function | Different tumors | [59,60] |
| Molecular vaccine & CAR-T | Increase CAR-T expansion | Solid tumors | [61] |
| Combined strategies to deal with solid-tumor challenges | |||
| OV19t & CD19-CAR-T | Deliver antigen and overcome heterogeneity | Solid tumors | [62] |
| OAd-TNFα-IL2 & CAR-T | Increase T-cell infiltration and reshape TME | PDA | [63] |
| OAd-expressing PD-L1 mini-body & HER2-CAR-T | Enhance CAR-T function | Prostate cancer | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, F.; Cui, J. CAR-T in Cancer Treatment: Develop in Self-Optimization, Win-Win in Cooperation. Cancers 2021, 13, 1955. https://doi.org/10.3390/cancers13081955
Guo F, Cui J. CAR-T in Cancer Treatment: Develop in Self-Optimization, Win-Win in Cooperation. Cancers. 2021; 13(8):1955. https://doi.org/10.3390/cancers13081955
Chicago/Turabian StyleGuo, Feifei, and Jiuwei Cui. 2021. "CAR-T in Cancer Treatment: Develop in Self-Optimization, Win-Win in Cooperation" Cancers 13, no. 8: 1955. https://doi.org/10.3390/cancers13081955