
Structural Bases for the Synergistic Inhibition of Human Thymidylate Synthase and Ovarian Cancer Cell Growth by Drug Combinations

 ,
,  ,
,  , , ,
, , , 
 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drugs and Chemicals
2.2. Protein Cloning and Purification
2.3. Crystallization and Structure Determination of the hTS:FdUMP:RTX Ternary Complex
2.4. Enzyme Kinetics and Inhibition Assays
2.5. Cell Lines
2.6. Cell Growth Assay
2.7. Biological Synergy Analysis
2.8. Cell Cycle Analysis
2.9. Statistical Analysis
3. Results
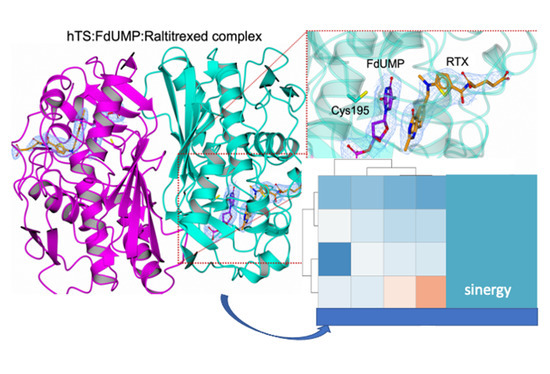
3.1. Crystal Structure of the hTS:FdUMP:RTX Ternary Complex
3.2. Synergistic Inhibition of the hTS Enzyme by FdUMP/RTX Combinations
3.3. Mechanistic Bases of the Synergism for Two Active-Site TS Inhibitors
3.4. Inhibitory Effects of RTX and 5FU Combinations on the Growth of Ovarian Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Li, X.; Qin, G.; Yang, Q.; Chen, L.; Xie, L. Biomolecular Network-Based Synergistic Drug Combination Discovery. Biomed Res. Int. 2016, 2016, 8518945. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.; Zhang, Z.; Ding, G.; Hui, L.; Li, Y.; Li, H. Personalized Anti-Cancer Drug Combination Prediction by an Integrated Multi-Level Network. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.-J.; Ladner, R.D. Standing the Test of Time: Targeting Thymidylate Biosynthesis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef]
- Avallone, A.; Di Gennaro, E.; Bruzzese, F.; Laus, G.; Delrio, P.; Caraglia, M.; Pepe, S.; Comella, P.; Budillon, A. Synergistic Antitumour Effect of Raltitrexed and 5-Fluorouracil plus Folinic Acid Combination in Human Cancer Cells. Anti Cancer Drugs 2007, 18, 781–791. [Google Scholar] [CrossRef]
- Gebbia, V.; Verderame, F.; Ferraù, F.; Bordonaro, R.; Callari, A.; Caruso, M.; Tirrito, M.L.; Valenza, R.; Cicero, G.; Borsellino, N.; et al. Raltitrexed plus Levofolinic Acid and Bolus/Continuous Infusion 5-Fluorouracil on a Biweekly Schedule for Elderly Patients with Advanced Colorectal Carcinomas. Ann. Oncol. 2006, 17, vii60–vii65. [Google Scholar] [CrossRef]
- Longo, G.S.; Izzo, J.; Chang, Y.M.; Tong, W.P.; Zielinski, Z.; Gorlick, R.; Chou, T.C.; Bertino, J.R. Pretreatment of Colon Carcinoma Cells with Tomudex Enhances 5-Fluorouracil Cytotoxicity. Clin. Cancer Res. 1998, 4, 469–473. [Google Scholar]
- Kano, Y.; Akutsu, M.; Suzuki, K.; Yazawa, Y.; Tsunoda, S.; Furukawa, Y. Schedule-Dependent Interaction Between Raltitrexed and 5-Fluorouracil in Human Colon Cancer Cell Lines In Vitro. Oncol. Res. 2001, 12, 137–148. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Tochowicz, A.; Pozzi, C.; Cardinale, D.; Ferrari, S.; Boum, Y.; Mangani, S.; Stroud, R.M.; Saxena, P.; Myllykallio, H.; et al. Hotspots in an Obligate Homodimeric Anticancer Target. Structural and Functional Effects of Interfacial Mutations in Human Thymidylate Synthase. J. Med. Chem. 2015, 58, 3572–3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuti, M.; Mangani, S. Crystallization of Soluble Proteins in Vapor Diffusion for X-Ray Crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef]
- Powell, H.R.; Johnson, O.; Leslie, A.G.W. Autoindexing Diffraction Images with IMosflm. Acta Cryst. Sect. D Biol Cryst. 2013, 69, 1195–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P. Scaling and Assessment of Data Quality. Acta Cryst. Sect. D Biol. Cryst. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Evans, P.R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Cryst. Sect. D Biol. Cryst. 2011, 67, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Cryst. Sect. D Biol. Cryst. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Cryst. Sect. D Biol. Cryst. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Phan, J.; Koli, S.; Minor, W.; Dunlap, R.B.; Berger, S.H.; Lebioda, L. Human Thymidylate Synthase Is in the Closed Conformation When Complexed with DUMP and Raltitrexed, an Antifolate Drug. Biochemistry 2001, 40, 1897–1902. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Cryst. Sect. D Biol. Cryst. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst. Sect. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Cryst. Sect. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. Validation of Protein Models Derived from Experiment. Curr. Opin. Struct. Biol. 1998, 8, 631–639. [Google Scholar] [CrossRef]
- Breitinger, H.-G. Drug Synergy—Mechanisms and Methods of Analysis. Toxic. Drug Test. 2012. [Google Scholar] [CrossRef] [Green Version]
- DiSaia, P.J.; Sinkovics, J.G.; Rutledge, F.N.; Smith, J.P. Cell-Mediated Immunity to Human Malignant Cells: A Brief Review and Further Studies with Two Gynecologic Tumors. Am. J. Obstet. Gynecol. 1972, 114, 979–989. [Google Scholar] [CrossRef]
- Andrews, P.A.; Murphy, M.P.; Howell, S.B. Differential Potentiation of Alkylating and Platinating Agent Cytotoxicity in Human Ovarian Carcinoma Cells by Glutathione Depletion. Cancer Res. 1985, 45, 6250–6253. [Google Scholar]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; IJcken, W.F.J.; van Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Lombardi, P.; Ferrari, S.; Monti, M.G.; Frassineti, C.; Costi, M.P. Modulation of the Expression of Folate Cycle Enzymes and Polyamine Metabolism by Berberine in Cisplatin-Sensitive and -Resistant Human Ovarian Cancer Cells. Int. J. Oncol. 2013, 43, 1269–1280. [Google Scholar] [CrossRef]
- Cho, Y.S.; Cho-Chung, Y.S. Antisense Protein Kinase A RIα Acts Synergistically with Hydroxycamptothecin to Inhibit Growth and Induce Apoptosis in Human Cancer Cells: Molecular Basis for Combinatorial Therapy. Clin. Cancer Res. 2003, 9, 1171–1178. [Google Scholar]
- Marverti, G.; Ligabue, A.; Guerrieri, D.; Paglietti, G.; Piras, S.; Costi, M.P.; Farina, D.; Frassineti, C.; Monti, M.G.; Moruzzi, M.S. Spermidine/Spermine N1-Acetyltranferase Modulation by Novel Folate Cycle Inhibitors in Cisplatin-Sensitive and -Resistant Human Ovarian Cancer Cell Lines. Gynecol. Oncol. 2010, 117, 202–210. [Google Scholar] [CrossRef]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the Biochemical Pathways of Thymidylate Synthase Perturbed by Anticancer Drugs: Novel Strategies to Overcome Cancer Chemoresistance. Drug Resist. Updates 2015, 23, 20–54. [Google Scholar] [CrossRef] [PubMed]
- Dolbeare, F.; Gratzner, H.; Pallavicini, M.G.; Gray, J.W. Flow Cytometric Measurement of Total DNA Content and Incorporated Bromodeoxyuridine. Proc. Nat. Acad. Sci. USA 1983, 80, 5573–5577. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.H.; Berger, F.G.; Lebioda, L. Effects of Ligand Binding and Conformational Switching on Intracellular Stability of Human Thymidylate Synthase. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2004, 1696, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.M.; Lovelace, L.L.; Lebioda, L. The R163K Mutant of Human Thymidylate Synthase Is Stabilized in an Active Conformation: Structural Asymmetry and Reactivity of Cysteine 195. Biochemistry 2008, 47, 4636–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almog, R.; Waddling, C.A.; Maley, F.; Maley, G.F.; Van Roey, P. Crystal Structure of a Deletion Mutant of Human Thymidylate Synthase Δ (7–29) and Its Ternary Complex with Tomudex and DUMP. Protein Sci. 2001, 10, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Guaitoli, G.; Tondi, D.; Luciani, R.; Henrich, S.; Salo-Ahen, O.M.H.; Ferrari, S.; Marverti, G.; Guerrieri, D.; Ligabue, A.; et al. Protein–Protein Interface-Binding Peptides Inhibit the Cancer Therapy Target Human Thymidylate Synthase. Proc. Nat. Acad. Sci. USA 2011, 108, E542–E549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, T.J.; Stroud, R.M. The Complex of the Anti-Cancer Therapeutic, BW1843U89, with Thymidylate Synthase at 2.0 å Resolution: Implications for a New Mode of Inhibition. Structure 1996, 4, 67–77. [Google Scholar] [CrossRef]
- Chen, D.; Jansson, A.; Sim, D.; Larsson, A.; Nordlund, P. Structural Analyses of Human Thymidylate Synthase Reveal a Site That May Control Conformational Switching between Active and Inactive States. J. Biol. Chem. 2017, 292, 13449–13458. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, C.; Ferrari, S.; Luciani, R.; Tassone, G.; Costi, M.P.; Mangani, S. Structural Comparison of Enterococcus Faecalis and Human Thymidylate Synthase Complexes with the Substrate DUMP and Its Analogue FdUMP Provides Hints about Enzyme Conformational Variabilities. Molecules 2019, 24, 1257. [Google Scholar] [CrossRef] [Green Version]
- Dowierciał, A.; Jarmuła, A.; Wilk, P.; Rypniewski, W.; Kowalska, M.; Frączyk, T.; Cieśla, J.; Rode, W. Mouse Thymidylate Synthase Does Not Show the Inactive Conformation, Observed for the Human Enzyme. Struct. Chem. 2017, 28, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems|Wiley; Wiley Classics Library; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1993; ISBN 978-0-471-30309-1. [Google Scholar]
- Fox, K.M.; Maley, F.; Garibian, A.; Changchien, L.-M.; Roey, P.V. Crystal Structure of Thymidylate Synthase A from Bacillus Subtilis. Protein Sci. 1999, 8, 538–544. [Google Scholar] [CrossRef]
- Hyatt, D.C.; Maley, F.; Montfort, W.R. Use of Strain in a Stereospecific Catalytic Mechanism: Crystal Structures of Escherichia Coli Thymidylate Synthase Bound to FdUMP and Methylenetetrahydrofolate. Biochemistry 1997, 36, 4585–4594. [Google Scholar] [CrossRef] [PubMed]
- Fritz, T.A.; Liu, L.; Finer-Moore, J.S.; Stroud, R.M. Tryptophan 80 and Leucine 143 Are Critical for the Hydride Transfer Step of Thymidylate Synthase by Controlling Active Site Access. Biochemistry 2002, 41, 7021–7029. [Google Scholar] [CrossRef] [PubMed]
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Jiang, Y.; Yang, Y.; Zheng, Y.; Zheng, J.; Jiang, H.; Zhang, S.; Lin, L.; Zheng, J.; Zhang, S.; et al. Determining the optimal 5-FU therapeutic dosage in the treatment of colorectal cancer patients. Oncotarget 2016, 7, 81880–81887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackman, A.L.; Kimbell, R.; Ford, H.E.R. Combination of Raltitrexed with Other Cytotoxic Agents: Rationale and Preclinical Observations. Eur. J. Cancer 1999, 35, S3–S8. [Google Scholar] [CrossRef]
- van der Wilt, C.L.; Smid, K.; Peters, G.J. Effects of Antifolates on the Binding of 5-Fluoro-2′-Deoxyuridine Monophosphate to Thymidylate Synthase. Biochem. Pharmacol. 2002, 64, 669–675. [Google Scholar] [CrossRef]
- Chen, X.; Ren, B.; Chen, M.; Wang, Q.; Zhang, L.; Yan, G. NLLSS: Predicting Synergistic Drug Combinations Based on Semi-Supervised Learning. PLoS Comput. Biol. 2016, 12, e1004975. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Hao, M.; Cheng, T.; Bryant, S.H.; Wang, Y. Predicting Drug–Drug Interactions through Drug Structural Similarities and Interaction Networks Incorporating Pharmacokinetics and Pharmacodynamics Knowledge. J. Cheminform. 2017, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Sidorov, P.; Naulaerts, S.; Ariey-Bonnet, J.; Pasquier, E.; Ballester, P.J. Predicting Synergism of Cancer Drug Combinations Using NCI-ALMANAC Data. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Ligabue, A.; Marverti, G.; Liebl, U.; Myllykallio, H. Transcriptional Activation and Cell Cycle Block Are the Keys for 5-Fluorouracil Induced Up-Regulation of Human Thymidylate Synthase Expression. PLoS ONE 2012, 7, e47318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Wilt, C.L.; Kuiper, C.M.; Peters, G.J. Combination Studies of Antifolates with 5-Fluorouracil in Colon Cancer Cell Lines. Oncol. Res. 1999, 11, 383–391. [Google Scholar]
- Genovese, F.; Gualandi, A.; Taddia, L.; Marverti, G.; Pirondi, S.; Marraccini, C.; Perco, P.; Pelà, M.; Guerrini, R.; Amoroso, M.R.; et al. Mass Spectrometric/Bioinformatic Identification of a Protein Subset That Characterizes the Cellular Activity of Anticancer Peptides. J. Proteome Res. 2014, 13, 5250–5261. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozzi, C.; Santucci, M.; Marverti, G.; D’Arca, D.; Tagliazucchi, L.; Ferrari, S.; Gozzi, G.; Losi, L.; Tassone, G.; Mangani, S.; et al. Structural Bases for the Synergistic Inhibition of Human Thymidylate Synthase and Ovarian Cancer Cell Growth by Drug Combinations. Cancers 2021, 13, 2061. https://doi.org/10.3390/cancers13092061
Pozzi C, Santucci M, Marverti G, D’Arca D, Tagliazucchi L, Ferrari S, Gozzi G, Losi L, Tassone G, Mangani S, et al. Structural Bases for the Synergistic Inhibition of Human Thymidylate Synthase and Ovarian Cancer Cell Growth by Drug Combinations. Cancers. 2021; 13(9):2061. https://doi.org/10.3390/cancers13092061
Chicago/Turabian StylePozzi, Cecilia, Matteo Santucci, Gaetano Marverti, Domenico D’Arca, Lorenzo Tagliazucchi, Stefania Ferrari, Gaia Gozzi, Lorena Losi, Giusy Tassone, Stefano Mangani, and et al. 2021. "Structural Bases for the Synergistic Inhibition of Human Thymidylate Synthase and Ovarian Cancer Cell Growth by Drug Combinations" Cancers 13, no. 9: 2061. https://doi.org/10.3390/cancers13092061
APA StylePozzi, C., Santucci, M., Marverti, G., D’Arca, D., Tagliazucchi, L., Ferrari, S., Gozzi, G., Losi, L., Tassone, G., Mangani, S., Ponterini, G., & Costi, M. P. (2021). Structural Bases for the Synergistic Inhibition of Human Thymidylate Synthase and Ovarian Cancer Cell Growth by Drug Combinations. Cancers, 13(9), 2061. https://doi.org/10.3390/cancers13092061